- Search Menu
- Advance Articles

Editor's Choice
- Information for authors
- Submission Site
- Open Access Options
- Why publish with the journal
- About DNA Research
- About the Kazusa DNA Research Institute
- Editorial Board
- Advertising and Corporate Services
- Journals Career Network
- Self-Archiving Policy
- Dispatch Dates
- Journals on Oxford Academic
- Books on Oxford Academic

Editor-in-Chief
Satoshi Tabata
About the journal
DNA Research is an internationally peer-reviewed journal which aims at publishing papers of highest quality in broad aspects of DNA and genome-related research …
Latest Articles

High-Impact Research Collection
Explore a collection of freely available high-impact research from 2020 and 2021 published in DNA Research .
Browse the collection

DNA Research is the official journal of Kazusa DNA Research Institute, published by Oxford University Press and supported by funding from Chiba Prefecture, Japan.

Why publish in DNA Research?
Growing Impact Factor, fully open access journal, low open access charges, and more.

Volume 26, Issue 6: TASUKE+: a web-based platform for exploring GWAS results and large-scale resequencing data
Read the Executive Editor’s commentary
Resource Articles: Genomes Explored

Email alerts
Register to receive table of contents email alerts as soon as new issues of DNA Research are published online.

Recommend to your library
Fill out our simple online form to recommend DNA Research to your library.
Recommend now

Committee on Publication Ethics (COPE)
This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE)
publicationethics.org
PubMed Central
This journal enables compliance with the NIH Public Access Policy Read more

- Open access
Open access options for authors.

Accepting high quality papers on broad aspects of DNA and genome-related research.
Related Titles

- Author Guidelines
Affiliations
- Online ISSN 1756-1663
- Copyright © 2024 Kazusa DNA Research Institute
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 23 January 2023
DNA synthesis technologies to close the gene writing gap
- Alex Hoose 1 ,
- Richard Vellacott 2 ,
- Marko Storch ORCID: orcid.org/0000-0003-1503-8282 3 , 4 ,
- Paul S. Freemont 3 , 4 &
- Maxim G. Ryadnov ORCID: orcid.org/0000-0003-4847-1154 1
Nature Reviews Chemistry volume 7 , pages 144–161 ( 2023 ) Cite this article
47k Accesses
48 Citations
111 Altmetric
Metrics details
- Synthetic biology
A Publisher Correction to this article was published on 30 June 2023
This article has been updated
Synthetic DNA is of increasing demand across many sectors of research and commercial activities. Engineering biology, therapy, data storage and nanotechnology are set for rapid developments if DNA can be provided at scale and low cost. Stimulated by successes in next generation sequencing and gene editing technologies, DNA synthesis is already a burgeoning industry. However, the synthesis of >200 bp sequences remains unaffordable. To overcome these limitations and start writing DNA as effectively as it is read, alternative technologies have been developed including molecular assembly and cloning methods, template-independent enzymatic synthesis, microarray and rolling circle amplification techniques. Here, we review the progress in developing and commercializing these technologies, which are exemplified by innovations from leading companies. We discuss pros and cons of each technology, the need for oversight and regulatory policies for DNA synthesis as a whole and give an overview of DNA synthesis business models.
Similar content being viewed by others

Synthetic DNA applications in information technology

Multiplexed CRISPR technologies for gene editing and transcriptional regulation

DNA-encoded chemical libraries
Introduction.
DNA is the information repository of life. Since its discovery, it has become an essential research tool for chemistry, biology and materials science. The past two decades have witnessed a remarkable progress in generating biological systems including viable microorganisms from synthetic genomes 1 , 2 . As a consequence of this success, the demand for DNA is increasing, driving the development of new technologies to provide DNA in greater purity, quantity and at a reduced cost 2 . These requirements have steered commercial priorities towards supplying synthetic DNA, as opposed to isolation of DNA derived from natural sources.
The ability to sequentially synthesize polynucleotides, nucleotide by nucleotide, allows for control over the composition and size of DNA. Synthetic DNA sequences provide researchers with a versatile tool to probe living systems, rather than relying on natural sequences isolated from organisms. Additionally, for some applications such as the amplification of inaccessible sequences, synthetic DNA is the only practical option. The development of DNA synthesis technologies may also be relevant in materials science and nanotechnology, for example, in DNA origami, to create new types of DNA architectures and functionalities using non-natural nucleotides or non-natural backbones, such as xeno nucleic acids (XNAs) 3 , 4 , 5 . Similarly, the synthesis of homo-polynucleotides, co-block and arbitrary polynucleotides has gathered momentum in applications in which single-stranded DNA acts as a scaffold or donor material for nanoscale devices or genome engineering 6 , 7 .
Innovations in next generation sequencing (NGS) have improved reading and editing DNA 8 and revolutionized cellular and populational genomic analysis, which are now applied in ‘mega-genomic’ initiatives 9 . DNA can be analysed at scale and low cost. However, the lack of large-scale DNA synthesis remains a barrier to technological advances and the large-scale analysis of genome structure and cellular function. This barrier highlights an existing gap between the well-developed ability to read DNA, identify and sequence genomes, with the less-developed ability to write DNA, and synthesize and produce DNA sequences of unlimited lengths and complexity.
In the current climate of DNA synthesis commercialization, businesses either offer DNA they synthesize themselves or ready-to-use automated synthesizers for researchers to make DNA in their own laboratories. Both routes make DNA synthesis accessible to those end-users who lack expert synthesis skills and as such ‘deskill’ DNA synthesis. However, widespread access to synthetic DNA through deskilling may lead to the misuse of synthetic DNA, which introduces the need for regulation to mitigate potential hazards resulting from the misuse 10 .
Here, we review both existing and emerging DNA synthesis technologies, with an emphasis on methodologies developed in industry as a means to accelerate the supply of long synthetic DNA. We also discuss challenges and opportunities that DNA synthesis brings for commercialization.
Why DNA — economy drive
Engineering biology holds promise for providing solutions to the global challenges of resource sustainability. Advances in engineering biology are already addressing industry needs by building effective partnership networks and investing into automation 11 , 12 . Indeed, 60% of all manufacturing inputs into the global economy could be produced biologically, whereas 30% of research and development spent is in biology-related industries 13 . DNA synthesis is indispensable in this regard as it provides the essence of engineering biology — DNA molecules of desired composition, complexity and length.
Four waves of DNA technologies
Following the sequencing of the human genome in the early 2000s 14 (the first wave of DNA technologies), the ability to ‘read DNA’ has advanced at a pace that has outstripped even Moore’s law, which predicts that the number of transistors doubles every 2 years 15 (Fig. 1a ). As this area matured, a second wave was driven by novel technologies such as de novo DNA synthesis and CRISPR gene editing, which has given an ability to ‘edit and write’ DNA 15 , 16 . This has enabled researchers to begin to ‘apply DNA’ by exploiting the abilities to read, edit and write DNA for products such as vaccines 17 , data storage 9 and drug delivery devices 6 or genome engineering to generate organisms with useful properties, such as heat-resistant plants 18 . The improved ability to apply DNA brings the need for synthesis on scale, to provide for industry challenges ranging from health security to environmental sustainability 17 , 18 , 19 . For instance, the COVID-19 pandemic has shown how quickly the rise in demand for vaccination can overwhelm existing production abilities. As RNA and DNA vaccines continue to get approved for major diseases, including COVID-19 (ref. 17 ), the demand for mass production of large DNA is growing. Similarly, synthetic DNA can be used in plants able to adapt to climate change, mitigating food security challenges 18 . Activated DNA repair increases the tolerance of plants to heat, while introducing synthetic genes make crops able to collect nutrients and water more efficiently in different conditions 18 , 19 . Effective DNA synthesis is therefore vital to close the gap between the ability to read and write DNA.

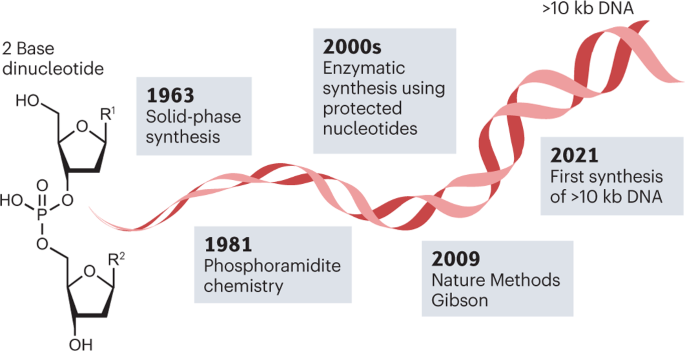
a , Productivity of DNA reading and DNA writing (synthesis) estimated in the number of nucleotides per person per day 15 . The grey arrow denotes the current gap in productivity between reading DNA and writing DNA. The dashed oval outline highlights the time frame within which the DNA synthesis industry achieved the majority of important milestones to close the gap. DNA synthesis data (red line) are available only for column-based synthesis instruments. The number of transistors per chip (Moore’s law) is shown for comparison. The graph uses the data available in the literature. b , Timeline of milestones in DNA synthesis technologies discussed in the report 20 , 25 , 26 , 34 , 35 , 37 , 42 , 71 , 146 . For simplicity not all milestones are shown. NTP, nucleoside 5′-triphosphate; PCA, polymerase cycling assembly; TdT, terminal deoxynucleotidyl transferase; TiEOS, template-independent enzymatic oligonucleotide synthesis. Copyright Wiley-VCH GmbH. Reproduced with permission from ref. 15 .
Since the structure of DNA was first understood 20 (Box 1 ), substantial milestones have been achieved, paving the way to a new industry. Over four decades, short but meticulous steps were taken to establish underpinning chemistry for the stepwise synthesis of DNA, nucleotide by nucleotide (Fig. 1b ). Chemical methods were developed to reliably provide short <200-nucleotide DNA chains, termed oligonucleotides. These methods were optimized for automatic synthesizers, which became indispensable tools for gene engineering and sequencing. Following this, the development of enzymatic and hybrid approaches to generate DNA that is longer and more complex than oligonucleotides has been achieved (Fig. 1b ). Companies have commercialized these approaches, offering services ranging from custom synthesis to benchtop DNA printers, making DNA synthesis accessible to non-expert users. This coincided with an apparent increase in ways that DNA can be applied while exposing the gap in DNA writing capabilities (Fig. 1a ). In recent years, DNAs of thousands of nucleotides in length have been produced, highlighting that this gap between the abilities to read and write DNA may close in the near future (Fig. 1b ).
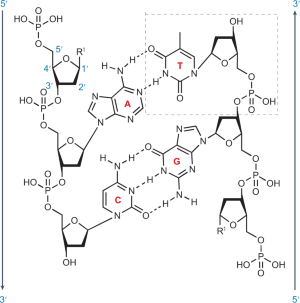
Box 1 Chemical structure of DNA
DNA is a monodisperse polymer of four building blocks, each comprising a base and a sugar residue, deoxyribose. Each block (adenosine (A), thymidine (T), guanosine (G), cytidine (C)) is coupled to a phosphate, which constitutes a nucleotide (thymidine highlighted by a dashed box). Nucleotides joined by phosphodiester bonds form a polynucleotide chain. The bases in this polymer pair up via three hydrogen bonds between G and C and two hydrogen bonds between A and T 200 . This selectivity, termed Watson–Crick base pairing, enables the formation of a double helix in which two complementary strands run anti-parallel to one another and intertwine with base pairs (A–T, C–G) stacked along the helical axis 20 . Owing to its self-complementarity, the double helix can be self-replicated from each of the two strands. Arrows indicate the 5′-to-3′ direction of the asymmetric ends.
Industry landscape for DNA synthesis
The DNA synthesis industry is rapidly growing, with an apparent shift towards greener solutions to reduce the dependence on chemical reagents and organic solvents with potentially adverse effects on the environment to mitigate their costly disposal routes 21 . New industry partnerships have been formed to introduce innovative technologies in the enzymatic DNA synthesis space. This can be exemplified by joint ventures between Codexis and Molecular Assemblies and between Integrated DNA Technologies and Danaher, which aim to advance enzymatic DNA synthesis abilities 21 , 22 , 23 . Promising technologies include plasmid template approaches, such as rolling circular amplification 24 , gene assembly approaches, such as Gibson assembly or polymerase cycling assembly (PCA) 25 , 26 , and template-independent enzymatic oligonucleotide synthesis (TiEOS), which exploits terminal deoxynucleotidyl transferase (TdT) as a DNA synthesis tool 27 , 28 .
A key challenge in DNA synthesis is the generation of >300-nucleotide DNA, which is limited by the elongation cycle efficiency, that is, the efficiency with which each nucleotide is incorporated in the sequence. For example, with the elongation cycle efficiency of 99%, the theoretical yield for an oligonucleotide comprising 120 nucleotides is ~30% (0.99 120 × 100%). However, for a 200 bp polymer/oligonucleotide, this is reduced to just 13%. Attempts to overcome this issue have focused on improving the accuracy and speed of DNA assembly processes. For commercial technologies, when >1 kb sequences are required, chemically produced >300 bp polynucleotides are used as building blocks for larger chains 29 , 30 , 31 . DNA printers, developed by some vendors, such as DNA Script, have enabled the parallel synthesis of multiple sequences, which can be linked together to produce longer chains. Other companies such as Molecular Assemblies focus on improving synthesis methodologies that might be implemented by developers of DNA printers or providers of synthetic genes.
Other vendors, such as ANSA Biotechnologies and Camena Bioscience, analyse the quality of the DNA they produce, to eliminate the need for the user to perform further sequencing or cloning. This has also allowed for more oversight to counter potential biosecurity risks. As synthetic DNA is involved in genetic engineering, there is a risk of its use in the production of pathogens and hence it is subject to an oversight or regulatory system. Similarly, companies that manufacture DNA printers use a cloud-based software enabling a degree of oversight for a desktop production mode. Table 1 provides examples of DNA synthesis companies highlighting pros and cons of their core technologies.
Making DNA: underpinning technologies
Over the past few decades, there has been significant interest in the development of DNA synthesis techniques. Starting with chemically synthesized dinucleotides 32 , de novo DNA synthesis was made possible and exploited in the process of deciphering the genetic code 33 . Advances in solid-phase synthesis inspired further synthetic improvements 34 , 35 , which led to the ground-breaking development of phosphoramidite chemistry for DNA synthesis in the 1980s resulting in the introduction of phosphoramidite oligonucleotide synthesis (POS) 36 , 37 .
Phosphoramidite synthesis
A typical solid-phase synthesis of oligonucleotides using phosphoramidite chemistry to build up a sequence, nucleotide by nucleotide, is given in Fig. 2A . This approach involves the stepwise addition of building blocks derived from 5′-protected dimethoxytrityl (DMT) nucleotide phosphoramidites 4 (refs. 38 , 39 , 40 ). This method was used by Applied Biosystems to develop the first automated DNA synthesizer in the 1980s, improving the accessibility of synthetic oligonucleotides 41 , 42 , 43 , 44 , 45 . Initial solid-phase methodologies used plastic or glass solid supports, onto which individual oligonucleotide sequences were chemically assembled 46 (steps c – i ). Since then, parallel in situ synthesis of oligonucleotides has been achieved using different microarray formats comprising multiple reaction sites, where one sequence is assembled onto one site, which can be controlled independently of other sites thus providing the synthesis of multiple sequences in a site-specific manner 47 , 48 , 49 , 50 .

A , Schematic representation of phosphoramidite solid-phase oligonucleotide synthesis. The method sequentially adds 5′-dimethoxytrityl-protected nucleoside phosphoramidites 4 upon activation by tetrazole to ensure sequence-specific strand elongation 35 , 36 , 37 , 38 , 39 . The steps of the process include a synthesis cycle comprising: protonation (step a ), detritylation (step b ), tetrazole activation and coupling (step c ), capping of unreacted nucleotides on the resin (step d ), oxidation (step e ), detritylation (step f ), which is repeated n times (step g ), and followed by cleavage from the support (step h ) and deprotection (step i ) to yield a desired DNA 10 . B , Mechanism of acid-catalysed depurination as a common side reaction in chemical synthesis 62 , 63 , 64 , 163 , comprising protonation (step j ), depurination (step k ), hydrolysis (step l ) and elimination (step m ). Resulting apurinic site 14 owing to the loss of a purine base (for example, adenine 13 ) is readily hydrolysed (steps c and d ) during the basic work-up steps required for removing base protecting group (PG).
Current technologies use silicon as a solid support onto which a million unique oligonucleotides can be written simultaneously 29 , 48 . Microscopic reaction clusters manufactured on a silicon chip decrease the reaction volume and significantly increase the output of DNA compared with single sequence synthesis methods 50 , whereas thermal control provides a means to monitor the incorporation of each nucleotide to enable site-specific DNA synthesis 51 , 52 .
However, the phosphoramidite method of DNA synthesis has drawbacks, including poor phosphoramidite bench stability, the need to use large quantities of organic solvents and the inability to synthesize poly-repeat sequences 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 . In addition, the acid required to remove the 5′-DMT protecting group (PG) can catalyse depurination (steps j–m in Fig. 2B ), a deleterious side reaction leading to the loss of purine bases (A, G) 13 from the synthesized DNA strand 12 , making this DNA strand susceptible to hydrolysis 14 and 15 and premature release 16 and 18 (refs. 62 , 63 , 64 , 65 ). As a result, depurination reduces the yield and purity of the desired oligonucleotide.
The workflow of oligonucleotide manufacturing, processing and purification is labour-intensive and remains largely the domain of service providers. Therefore, synthesis capabilities have become centralized within specialist reagent manufacturers. Leading vendors such as Agilent Technologies, GenScript, Integrated DNA Technologies, ThermoFisher, TriLink, Dharmacon, Twist Bioscience and others produce custom DNA (and RNA) on demand in a range of formats. For those users who wish to decrease the lead time for such services, there is a range of instruments, for example, Cytiva’s ÄKTA oligonucleotide synthesizers, which can be purchased and operated on a daily basis.
Traditionally, molecular biology relied on short DNA sequences, such as primers for PCR or probes for molecular detection, amplification and modification applications. More recently, asymmetric PCR methods 66 have enabled advances in the amplification of individual DNA strands of thousands of nucleotides in length 67 , 68 . Now researchers seek longer sequences of varied composition including entire genomes with a single-base accuracy, which must be assembled from scratch 31 , 69 . Such long sequences are incompatible with the phosphoramidite method whose efficacy in the synthesis of pure DNA reduces beyond approximately 200-bp oligonucleotide sequences.
To synthesize long DNA, the elongation cycle efficiency must be increased to improve yields, and the incomplete removal of PGs and side reactions such as depurination must be minimized or avoided 62 , 70 . Longer sequences must be assembled from smaller strands in error-correcting stages using an alternative methodology. Enzymatic approaches are most attractive in this regard and are also scalable, stereospecific and environmentally friendly 21 . Enzymes can mediate mismatch recognition enabling the selective annealing of complementary strands, reduce the number of steps in each elongation cycle by eliminating the need for coupling reagents and decrease the dependence on organic solvents. Enzymes can promote synthesis with or without DNA templates, through amplification or in the synthesis of de novo sequences.
Enzymatic oligonucleotide synthesis
Enzymatic synthesis uses the principles of solid-phase synthesis. A short strand of DNA synthesized on a solid support can be extended by DNA polymerases using nucleoside 5′-triphosphates (NTPs) 27 . DNA polymerases use a template DNA strand that provides base pairing, thereby selecting the incoming nucleotide. This means that although polymerases are effective in amplifying existing DNA templates, they are unable to generate de novo DNA sequences. Therefore, an alternative enzyme is required to efficiently elongate polynucleotide chains in the absence of a template strand 71 , 72 , 73 . Such a polymerase has been identified as TdT and is integrated into commercial TiEOS methods 27 , 28 , 71 (Fig. 3A ).

A , Schematic representation of 3′-protected nucleoside 5′-triphosphate (NTP) approach. Resin beads are pre-loaded with an initiator DNA (iDNA) 19 to provide a template for binding of terminal deoxynucleotidyl transferase (TdT) and as a post-synthesis cleavage site 27 , 72 , 77 , 78 , 95 . Oligonucleotide synthesis then proceeds in a stepwise fashion in the 5′-to-3′ direction. TdT ligates NTP 20 to the 3′ terminus of the growing oligonucleotide chain with each NTP protected at 3′-OH with a protecting group (PG) 24 – 26 (refs. 27 , 28 , 82 ). The resin is washed to remove surplus reagents and the pyrophosphate by-product of the ligation. After deblocking or deprotection of the 3′-PG (step b ), the resin-bound 3′-OH nucleophile of 22 becomes available for the next synthesis cycle (step c ). The complete sequence is assembled by repeating the cycle of TdT-catalysed NTP(PG) coupling (step a ) and deblocking (step b ). On completion, the synthesized oligonucleotide 4 is cleaved from the solid support (step d ) by uracil DNA glycosylase. B , Examples of NTP(PG)s used in the method — 3′-azidomethyl-protected NTPs 24 by Nuclera Nucleics, Molecular Assemblies, 3′-ONH 2 -protected NTPs 25 by DNA Script and 3′-O-2-nitrobenzyl 26 by Camena Bioscience 97 , 100 , 101 , 108 , 118 . C , Schematic representation for alternative (tethered) protecting strategies. 3′-Unprotected NTPs (cytidine) are supplied pre-immobilized within the TdT-active site 28 , via a short and labile linker 121 , 129 . TdT then catalyses the incorporation of this NTP into the growing DNA strand 30 (step a ) and sterically prevents the uncontrolled polymerization of the NTP until the linker is cleaved (step b ), releasing the oligonucleotide 32 . The cycle is repeated (step c ) until the desired oligonucleotide 33 is completed (step d ). Asp, aspartic acid; DTT, dithiothreitol; TCEP, tris-carboxyethylphosphine; TiEOS, template-independent enzymatic oligonucleotide synthesis.
Template-independent enzymatic oligonucleotide synthesis
TdT elongates oligonucleotides in the 5′-to-3′ direction in a promiscuous manner, accepting any of the four canonical nucleotides, resulting in the concomitant formation of different sequences 73 . An effective solution is to control the incorporation of nucleotides via a ‘reversible termination’ mechanism (Fig. 3A ). This mechanism uses NTPs modified with a synthesis-interrupting ‘terminator’ or PG at the 3′ position, which ensures the addition of a single nucleotide per reaction step and is subsequently removed to incorporate the next desired nucleotide 24–26 (refs. 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 ). To this effect, TiEOS uses resin beads pre-loaded with a chemically synthesized single-stranded initiator DNA 19 (refs. 72 , 83 ), onto which TdT ligates 3′-protected NTPs 20 into a desired sequence 23 . At each step of elongation cycle, steps a and b (Fig. 3A ), a washing step is used to remove side-products and surplus reagents, and the deblocking of 3′-PG is performed at the end of each cycle before the next elongation cycle (steps b and c in Fig. 3A ). Initiator DNA 19 incorporates a highly specific deoxyuridine cleavage site at its 3′ end which is enzymatically labile. This site is cleaved by uracil DNA glycosylase upon the completion of the synthesis to release the assembled sequence 23 from the resin 27 , 74 , 75 , 76 , 77 , 78 .
TdT methodologies as a main paradigm in DNA synthesis
As more companies test this approach, several important and unique limitations of TdT have been reported. First, the enzyme demonstrates a preference for the incorporation of some nucleotides over others 84 . This bias could increase the rates of sequence-specific errors. Second, TdT works only on single-stranded DNA. This is attributed to a lariat-like loop in the enzyme, which acts as a steric shield that prevents a double-stranded DNA template accessing the active site of the enzyme 71 , 80 . Consequently, the efficiency of the synthesis is reduced if the strand under construction begins to form secondary structures 83 . Third, like all DNA polymerases, TdT-catalysed phosphoryl transfer requires divalent cations to synthesize DNA from NTPs 85 . However, unlike other DNA polymerases, which typically require Mg 2+ to catalyse the synthesis of DNA molecules, TdT can use various divalent metal cations, for example, Co 2+ , Mn 2+ , Zn 2+ and Mg 2+ , with the NTP incorporation tailored by the cation identity. For instance, the use of Mg 2+ favours the incorporation of deoxyguanosine triphosphate and deoxyadenosine triphosphate, whereas Co 2+ promotes the incorporation of deoxycytidine triphosphate and deoxythymidine triphosphate 84 , 85 , 86 . Crucially, this bias extends to protected NTPs used in DNA synthesis 27 , 28 , 80 , prompting researchers to develop methods to mitigate the bias 87 . Additional features of TdT, which impact on the choice of PGs and synthesis efficiency, include the DNA phosphorylation capacity and phosphatase activity of the enzyme 88 , 89 , 90 . Thus when a growing oligonucleotide chain is exposed to a mixture of NTPs, TdT would preferentially incorporate certain nucleotides resulting in the synthesis of homopolymeric chains of varying lengths.
To address these shortcomings, different approaches are being explored. By analogy to peptide synthesis 91 , microwave irradiation can be tailored to accelerate synthesis using DNA polymerases that work on double-stranded DNA or convert a desired double-stranded DNA into its single-stranded form, which is accessible to TdT 92 , 93 .
To avoid the random incorporation of NTPs into a growing DNA chain by TdT, suitable 3′-PGs have been developed for NTPs 24– 26 (Fig. 3B ), which facilitate a sequential synthesis cycle comprising 3′-PG deblocking, resin washing and the coupling of NTPs (Fig. 3A ). This cycle constitutes a technologically optimized TiEOS that is already adopted by several companies such as DNA Script and Nuclera Nucleics 23 , 94 , 95 .
Important optimizations for this approach concern the design of 3′-PGs, for example, DNA Script chooses 3′-ONH 2 -protected NTPs 25 (refs. 94 , 96 ), whereas Nuclera Nucleics and Molecular Assemblies prefer azidomethyl terminators 24 (refs. 97 , 98 , 99 , 100 ) and Camena Bioscience appears to favour 2-nitrobenzyl 26 as a 3′-PG 101 . Other PGs are attempted for the protection of 3′-OH and the bases of NTPs 102 , 103 , 104 , 105 , 106 , with parallel efforts focusing on PGs for XNA synthesis 90 , 107 . However, TdT must be able to accommodate the protected nucleotides in its active site, which limits the choice of PGs or requires the re-engineering of the enzyme for compatibility with 3′-PG. Indeed, 3′-PG NTPs are not natural substrates for TdT owing to the steric hindrance in the active site of the enzyme 71 , and their development is closely guarded by vendors 82 , 101 , 108 . Re-engineering of TdT may provide a solution to this issue and also may aid the development of thermostable TdT 81 , 95 , 109 , 110 , 111 , 112 , 113 , 114 . DNA Script, Nuclera Nucleics and Molecular Assemblies are active players in this area 76 , 77 , 98 , 109 , 115 , 116 , 117 , 118 , 119 , 120 , 121 , 122 , 123 , 124 , 125 , 126 , 127 , 128 , whereas Camena Bioscience has developed a proprietary combination of high-fidelity enzymes to achieve template-free DNA synthesis 101 , 128 (Table 1 ).
Other companies adapt an alternative approach to temporarily cap the growing oligonucleotide chain by developing 3′-OH protecting strategies. For example, Molecular Assemblies furnish incoming NTPs with blocking groups to sterically shield its 3′-OH from elongation until removal 129 , 130 , 131 . In another strategy, ANSA Biotechnologies tether TdT to the base of an incoming NTP via a cleavable linker to prevent the formation of homopolymeric nucleotide tracts 132 (Fig. 3C ). The α-phosphate group of the NTP 28 reacts with the 3′-OH of the growing oligonucleotide 27 , whereas its unprotected 3′-OH remains sterically shielded by the enzyme 30 , which prevents polymerization 121 . Cleaving the linker releases TdT 31 and the elongated oligonucleotide 32 . By repeating the cycle, steps a–c , the desired sequence can be assembled and released 33 .
The yield, purity and achievable lengths of chemically synthesized oligonucleotides depend on the effective completion of each coupling cycle. Although a two-step cycle used in TiOES is an improvement to the four steps required in POS, TiEOS is unlikely to provide the cost-effective and time-effective synthesis of full-length genes. A nearly quantitative elongation cycle efficiency of 99.9% results in a <37% yield for a 1,000-bp (or 1 kb) DNA strand. By contrast, the 99.7% efficiencies reported by DNA Script would result in yields less than 5% 23 . However, even with 99.9%, a yield of <5% for 3 kb DNA would be achievable. For example, Camena Bioscience applied their proprietary de novo synthesis and gene assembly technology — gSynth — for the construction of a 2.7 kb plasmid vector, pUC19 (refs. 101 , 128 , 133 , 134 ). As the synthesis progresses, >3 kb polynucleotide chains can form stabilized secondary structures (for example, hairpins) with detrimental effects on the elongation cycle efficiency 83 , 135 . Microwave treatments might mitigate this issue, but still within the 3 kb range 92 .
Despite limitations, TiEOS reduces the complexity of crude oligonucleotides by minimizing the number of possible impurities, uses ‘green’ reagents, and relies on fewer steps per synthesis cycle when compared with POS. These benefits of TiEOS enhance product purity and quality compared with POS, but still do not achieve quantitative elongations or resolve the detrimental impact of secondary structure formation on DNA synthesis 83 , 135 . Therefore, TiEOS is viewed as a promising ‘green’ methodology for the synthesis of <3 kb DNA. To synthesize larger constructs (that is, gene clusters or chromosomes), TiEOS may be used to generate shorter fragments that can then undergo ligation by Gibson assembly or PCA.
Technologies for DNA of unlimited length
The complementary nature of DNA (Box 1 ) and the wealth of enzymes capable of polymerizing, cleaving, nicking, ligating and mutating DNA have resulted in the development of various assembly methods. By improving enzymes and assembly standards, the accuracy and number of DNA molecules that can be combined in a single step have improved, which has been applied in the synthesis of a minimal bacterial genome 136 and synthetic yeast chromosomes 69 . With DNA assembly methods reviewed in detail elsewhere 137 , 138 , here we focus on two essential methods for DNA synthesis workflows, namely, Gibson assembly 25 , 139 , 140 and PCA 26 , 141 .
Gibson assembly
Gibson assembly is an enzymatic approach used to complement POS and TiEOS methods 25 , 139 . Although this approach is inefficient for the synthesis of short strands (<100 nucleotides) 142 , it is used to assemble large DNA fragments 143 , 144 , 145 (Fig. 4A ). Gibson assembly starts with two DNA duplexes 34 and 35 , which have complementary terminal overlap regions. Each strand of these DNA duplexes is degraded by an exonuclease from the 5′-end, generating the 3′-‘sticky’ ends of duplexes 36 and 37 . The sticky ends of these two duplexes are then annealed in step b and repaired by a polymerase, which adds missing nucleotides to the two strands using base pairing interactions. A DNA ligase then stitches the nucleotides of each strand together to form the desired duplex product 38 .

A , Gibson assembly 25 , 139 , 140 . Two duplex DNA strands 34 and 35 are selected with a complementary terminal overlap region (black). Digestion with T5 exonuclease (step a ) degrades each strand of the DNA duplexes in the 5′-to-3′ direction, yielding the sticky ends of 36 and 37 , followed by the annealing of complementary sticky ends between the two DNA duplexes 143 (step b ). Phusion polymerase and Taq ligase are then combined (step c ) to ligate the two short DNA duplexes into a single, long DNA duplex construct 38 (refs. 144 , 145 ). The process is repeated (step d ) for gene assembly. B , Polymerase cycling assembly (PCA) 26 , 141 . High-purity synthetic oligonucleotides 39 are designed, such that annealing of complementary overlaps generates the desired long duplex DNA construct (step a ). The desired construct is then assembled in either a single step from 43 or two steps from 39 using a DNA polymerase (step b ) to yield template 40 . PCR amplification (steps c and d ) of 41 amplifies the desired long duplex DNA construct 42 .
Multiple rounds of Gibson assembly yield large genetic fragments for a range of applications, such as protein expression to transcriptional control. However, the process remains laborious. A non-automated gene assembly is time-consuming, which is compounded by the need for high-purity oligonucleotides in large quantities. Oligonucleotide purity is also critical to ensure correct assembly, even small percentages of deletions can create substantial frameshift mutations within the open reading frame of a desired DNA — the section of DNA that is transcribed by enzymes into RNA. Even a single deletion can shift the reading frame compromising RNA transcription, which renders the DNA unusable. Because of this, the final gene products are cloned into plasmids and transformed into bacterial strains to confirm the presence of the desired DNA sequence. The synthesis of longer genes often requires multiple cloning and repeated Gibson assembly steps causing additional costs and long lead times.
Polymerase cycling assembly
Owing to the development of PCR, the amplification and sequencing of DNA are now routine 146 , 147 , 148 . Watson–Crick base pairing was used in conjunction with PCR to develop a method for stitching together pools of synthetic oligonucleotides in a technology termed PCA 26 . In PCA, target oligonucleotides, which are referred to as ‘sense’, are annealed via complementary overhangs to oligonucleotides corresponding to a complementary, antisense strand (Fig. 4B ). Each oligonucleotide, with the exception of those positioned at the 5′ termini of each strand, hybridizes with two complementary oligonucleotides within the opposite strand. This produces an annealed construct 39 and 43 , with alternating ‘gaps’ present within the sense and antisense strands. The gaps are then filled in using a polymerase to generate a duplex DNA template 40 and 41 for PCR amplification. Following this assembly phase, external primers, which are complementary to the 5′ ends of the duplex DNA template, are introduced to perform a PCR reaction, which amplifies the target sequence to yield the final product 42 . Using this approach, a plasmid of >2.5 kb has been produced from short, chemically synthesized oligonucleotides 26 .
Like for Gibson assembly, the performance of PCA can be compromised by impurities of synthetic oligonucleotides. Other disadvantages of PCA include the dependence of the method on sequence confirmation from an individual clone and reliance on high-fidelity proof-reading PCR enzymes which must be used to copy constructed genes to prevent mutations during amplification. Yet, owing to the limitations on the length of iteratively synthesized polynucleotides, Gibson assembly and PCA remain the main practical options for making large DNA.
Emerging commercialized technologies
Companies developing novel ways to make DNA focus on meeting one of two main requirements: longer DNA sequences or greater numbers of DNA constructs made in parallel. Both templated and template-independent approaches are developed for large-scale production and the assembly of long DNA. Increasingly, companies place an emphasis on DNA synthesis services, which remain highly competitive and necessitate tighter control over the distribution of synthetic DNA. Automation offers opportunities to minimize expert involvement in DNA synthesis and is being realized by the supply of benchtop DNA printers. Typically, industry tailors synthetic methods for specific DNA targets, in terms of both complexity and length. This is driven by challenging and topical applications such as the synthesis of DNA vaccines or gene therapeutics. These applications demonstrate the value of providing DNA products in high yield and purity. A number of exciting developments in industry are discussed subsequently to exemplify the progress in the field of DNA synthesis.
Thermally controlled synthesis
A progressive solution to parallel DNA synthesis, proposed by Evonetix, is thermally controlled synthesis. This method is compatible with both phosphoramidite and TiEOS approaches 29 , 51 , 52 , 149 , 150 and offers the synthesis of DNA libraries, with sequences immobilized on discrete thermally controlled reaction sites of silicon chips. Thermal heating allows to selectively cleave PGs (5′ for phosphoramidite or 3′ for TiEOS, step a in Fig. 5 ), from the termini of specific reaction sites for elongation 47 . The entire chip can then be exposed to a TiEOS or phosphoramidite elongation cycle, selectively elongating only oligonucleotides immobilized on the heated reaction 49 . Unheated sites retain their thermally labile terminal PGs rendering these chains unavailable for elongation 46 and 48 (refs. 150 , 151 , 152 ) (Fig. 5 ).

Terminally protected oligonucleotide strands 44 and 45 are immobilized on discrete reaction sites (sites 1 and 2). Thermal heating of a chosen site (site 2) cleaves terminal protecting groups (PGs) 47 (step a ), enabling selective elongation of strands on this site 29 , 51 , 52 , 151 (steps b and c ) Elongation of a desired oligonucleotide is performed via TiEOS or the phosphoramidite method to selectively generate 49 (refs. 27 , 37 ). Repeated steps a– c on other selected reaction sites (for example, site 1) sequentially produce bespoke oligonucleotides 50 (steps d and e ). Thermally assisted reagent treatment of selected sites cleaves safety catch linkers and liberates oligonucleotides 53 from a chosen site 29 , 151 (step f ). The liberated oligonucleotides (step g ) anneal to complementary chip-bound oligonucleotides 52 producing perfectly annealed double-stranded DNA 54 , which has a higher denaturation temperature than that of DNA duplexes formed by oligonucleotides with mismatches 55 . The heating of reaction site 1 allows the mismatched oligonucleotides 56 to be washed away 150 (step h ). Thermally assisted reagent treatment of site 1 cleaves safety catch linkers and releases the desired duplex DNA 57 from the chip 29 , 151 (step i ). This liberated DNA is annealed to a chip-bound complementary DNA duplex 58 to form the nicked construct 59 (step j ). The process is repeated to elongate the double-stranded DNA until the desired gene is assembled.
As with other DNA synthesis approaches, the elongation cycle efficiencies are the limiting factor. In each reaction site, a percentage of insufficient thermolysis of PGs is expected. With every elongation cycle, deletion sequences would accumulate creating impurities similar to the desired product. Evonetix addressed this issue by tethering each immobilized oligonucleotide to the chip via a linker that is labile to thermally assisted chemical cleavage. Once the desired strands 53 are assembled, the site on the chip to which they are immobilized is heated resulting in the cleavage of the linker and the liberation of these strands into solution. These liberated strands can be made complementary to oligonucleotides 52 , which remain immobilized on the chip and can be subsequently annealed together to yield double-stranded DNA molecules 54 (ref. 150 ). Any imperfectly annealed oligonucleotide pairs 55 , for example, owing to truncated sequences, can be thermally denatured at lower temperatures than the desired DNA 54 . Such a process of thermal purification removes incorrect sequences 56 , yielding a double-stranded DNA product with the desired base pairing 57 (refs. 150 , 152 ). If these duplex DNA pairs have sticky ends complementary to strands 58 immobilized on another site of the chip, then sequential pairs can be annealed into a ‘nicked’ construct 59 . Repetition of this process yields a double-stranded product, the length of which is virtually unlimited. The nicks present in the strand could be repaired by a DNA ligase into the double-stranded DNA of a desired length and the construct may be amplified by PCR. Evonetix is anticipated to offer desktop DNA printers based on this technology. These plug-and-play instruments will feature user interfaces and design algorithms implemented in the cloud to enable control over biosecurity of gene synthesis 52 .
Gene synthesis from libraries
Ribbon Biolabs has developed a convenient synthesis of long (>10 kb) duplex DNA, using the convergent assembly of double-stranded oligonucleotide pools 153 (Fig. 6A ). The methodology requires the synthesis of a library of tens of thousands 5′-phosphorylated single-stranded oligonucleotides of high purity and 8–26 nucleotides in length, encompassing all the necessary building blocks for DNA synthesis 154 . Each oligonucleotide has a designated 5′-phosphorylated reverse complement strand in the library, with annealing overhangs of four nucleotides designed for each strand at the 5′-end. The assembly process requires the denaturation and annealing of pair of complementary oligonucleotides 60 and 61 and 62 and 63 to generate a library of duplex DNA constructs 64 and 65 , each with two four-nucleotide sticky ends at the 5′ termini of both strands 153 .

A , Gene synthesis from diverse oligonucleotide libraries. 5′-Phosphorylated sense oligonucleotide strands 60 and 61 are annealed (step a ) to complementary 5′-phosphorylated antisense strands 62 and 63 . The resulting DNA duplexes 64 and 65 have two 5′-overhangs or sticky ends that are used to anneal (step b ) the duplexes into an extended, ‘nicked’ duplex 66 (‘nick’ highlighted in magenta). T4 DNA ligase is then used (step c ) to stitch the oligonucleotides at the nick site into an elongated, larger DNA duplex 67 . Cycles of annealing and ligation are repeated until the desired gene is assembled 153 , 154 , 155 (step d ). B , DNA microarrays 30 . A library of bespoke single-stranded oligonucleotides 68 is generated with 3′-terminal and 5′-terminal DNA ‘barcodes’ on a miniaturized chip 30 , 47 , 48 , 50 , 164 , 166 , 167 (step a ). These sequences are cleaved (step b ) from the microchip to yield a pool of template oligonucleotides with a range of DNA ‘barcodes’ (only two, black and brown, ‘barcodes’ are shown for clarity) 164 , 167 . Primers selectively anneal to either the ‘brown’ 69 or ‘black’ 70 DNA barcodes and specifically amplify oligonucleotides via PCR (step c ), according to the identity of the barcode at its 3′ and 5′ termini 146 , 147 , 148 . The resulting duplex DNA constructs 71 and 72 still contain the DNA barcodes at their termini, which must be removed prior to gene assembly. DNA barcodes are cleaved (step d ) from the duplex DNA 71 and 72 by type IIS restriction endonucleases (REN), giving rise to assembly pools of sequences 73 and 74 with sticky ends 30 . Duplex DNA fragments are annealed (step e ) via complementary sticky ends and assembled into desired genes 75 and 76 via Gibson assembly 25 , 139 , 140 . C , Rolling circle amplification (RCA) 175 . Template plasmid DNA 77 with a desired gene cassette (green) and protelomerase sites (magenta) is thermally denatured (step a ) to create a single-stranded template 78 (ref. 178 ). A complementary primer binds to the protelomerase sites 79 (step b ) and the template is amplified via RCA (steps c–e ), to produce double-stranded concatemeric DNA 82 with alternating copies of the desired cassette (green) and the unwanted plasmid backbone (black) 176 , 181 , 182 , 183 . Protelomerase then cuts (step f ) the duplex at its recognition sites and ligates the cut ends generating covalently closed ‘doggybone’ DNA (dbDNA) 84 and a circular plasmid DNA 83 as a by-product. The circular backbone of the plasmid DNA is subsequently cut (step g ) by REN and digested (step h ) by exonucleases 179 .
Duplex DNA fragments 64 and 65 with 5′-phosphorylated, four-nucleotide sticky ends are then annealed in step b and ligated in step c together in a convergent synthesis giving rise to larger duplex DNA 67 for further assembly. Repeated cycles of the annealing and ligation of these building blocks give the desired duplex construct 155 . Terminal duplex DNA blocks have a single ‘blunt end’ and a single ‘sticky end’ to yield linear duplex DNA products. Once the final DNA duplex is obtained, it can be amplified by PCR using a high-fidelity polymerase to provide product yield for the customer. This technology from Ribbon Biolabs is therefore analogous to a convergent Gibson assembly approach 25 .
Gene synthesis from DNA microarrays
A similarly effective approach has been developed by Twist Bioscience through miniaturizing and performing gene synthesis onto a silicon microarray chip 47 , 48 , 49 , 50 . A grid of 25,000 discrete reaction sites is generated using an ink jet printing. Specialist reagents are then delivered to each site. The method enables the selective elongation of several desired sequences out of a library of tens of thousands with improved elongation efficiencies (Fig. 6B ). Low concentrations and volumes used in the method (approximately femtomole) permit starting reagents to be used in a large excess, whereas the acidic 5′ detritylation solution is neutralized by basic oxidation to prevent depurination 47 , 156 , 157 , 158 , 159 , 160 , 161 , 162 , 163 . Assembled sequences are produced in relatively low quantities, which necessitates the use of PCR to generate sufficient DNA for gene assembly 30 . Microarrays provide complex pools of DNA 68 , which can include both strands of a complementary duplex. After annealing, the duplex is used for template-specific 69 and 70 amplification by PCR to generate larger quantities of selected sequences 71 and 72 , respectively, which hybridize efficiently with primers 164 , 165 , 166 , 167 . In this format, duplex DNA can be selectively amplified from a complex pool of sequences in parallel. The assembly subpools of double-stranded DNAs 71 and 72 amplified by PCR are then digested by type IIS restriction endonucleases to generate sticky-ended duplexes 73 and 74 , respectively. These duplexes are then used as building blocks for Gibson assembly to assemble desired genes 75 and 76 at a fraction of the cost of traditional column synthesized oligonucleotides 30 , 168 . Miniaturization has benefitted other areas too. Notably, microarrays have proven instrumental for optimizing the parallel synthesis of oligonucleotides on TiOES platforms, including the impact of initiating strands and chemically modified NTPs on enzymatic DNA synthesis. Microarrays have also prompted early considerations for DNA nanofabrication, synthesis multiplexing and compatibility of enzymes with alternative polymerization methods 169 , 170 , 171 , 172 .
Rolling circle amplification technique
Touchlight Genetics has commercialized a technology to scale up the manufacturing of large DNA using a linear closed ‘doggybone’ DNA (dbDNA), named after its structure 84 resembling a doggy bone (Fig. 6C ). dbDNA is produced via rolling circle amplification from a plasmid template 77 (refs. 173 , 174 , 175 , 176 ). The template must be engineered to contain desired expression cassettes, for example, inverted terminal repeats, directly between two 28 nucleotide protelomerase recognition sites 177 , 178 , 179 . The denatured plasmid template 78 is amplified by DNA polymerase, in the presence of a primer that binds to the protelomerase recognition sites 79 . Once the plasmid has been replicated, the DNA polymerase continues to repeatedly replicate the plasmid template via rolling circle amplification, displacing any pre-existing synthesized strands from the template 81 . The polymerase then binds to these liberated single strands of DNA and replicates the complementary strand to generate concatemeric double-stranded DNA 82 . Protelomerase is added to generate a double-stranded DNA break and form a hairpin loop to re-seal the ends resulting in dbDNA 84 and a circularized plasmid DNA by-product 83 (refs. 179 , 180 , 181 , 182 , 183 ).
Restriction endonucleases are carefully selected such that they can digest the unwanted plasmid backbone 83 , but their restriction sites are not present in dbDNA 84 (ref. 180 ). Digestion of the products of the reaction liberates dbDNA 84 and the undesired linearized plasmid backbone 85 . A subsequent digestion of this mixture with exonucleases produces a mixture of nucleotides, enzymes and buffers, which can be readily separated from the desired product. dbDNA 84 is then purified to provide a minimal, linear DNA vector encoding virtually any long sequence of interest. These sequences can be either complex or unstable and can be re-amplified using the same process to rapidly generate multigram quantities of large DNA free of bacterial or endotoxin contaminations 176 . As a manufacturing platform, this approach permits the production of large DNA five times faster than traditional fermentation methods (Table 1 ).
Business models and deskilling
Approaches combining chemical and enzymatic syntheses, sequence selection and assembly are set to undergo continuous development. However, as the underpinning chemistry for synthetic DNA is unlikely to change markedly, the elongation cycle efficiency remains the main limiting factor. This has prompted companies to develop complementary capabilities such as highly parallelized, miniaturized and automated synthesis, while promoting user autonomy in producing DNA (Fig. 7 ).

DNA synthesis technologies plotted versus complexity levels of the current offerings (left axis) by different companies who develop these methods and versus user autonomy (deskilling) that these companies offer (right axis), from expert involvement to complete autonomy for the end user. For example, template-independent enzymatic oligonucleotide synthesis (TiOES) is the basic technology for DNA Script who offer customized and multiple sequences, while integrating automation into their synthesis workflows, which results in the development of benchtop DNA printers that the end user can buy and use with a minimum expert involvement. Gibson, Gibson assembly approaches; POS, phosphoramidite oligonucleotide synthesis; RCA, rolling circle amplification.
Automation and services
Research focuses on improving the synthesis of DNA sequences in parallel. This is known to increase the probability of errors, in particular for sequences that are difficult to amplify, such as repeat or GC-rich sequences. Automation provides a compelling direction. Companies exploit advances in other areas, such as electronics and microfluidics to improve DNA synthesis. This has aided the identification and removal of errors, increased accuracy, scale and speed to a far greater extent than non-automated approaches 29 , 51 , 52 . For example, Evonetix developed a platform for high fidelity and rapid gene synthesis, which is controlled by electrochemical processing of each of many thousands of independent reaction sites on a silicon chip, in a highly parallelized fashion. The combination of parallel synthesis and site-specific thermal control has the potential to address limitations of difficult sequences. For instance, sequences with a high GC content, which require higher melting temperatures than other sequences and can form stable secondary structures, can be synthesized at elevated temperatures. However, when using such temperatures, high site specificity is necessary to prevent mis-annealing (Table 1 ).
With several enzyme companies now active in this space, various business models have emerged with an increasing emphasis on DNA assembly and benchtop printers. For example, the size of a DNA Script’s Syntax instrument offered as a benchtop DNA synthesizer is similar to that of a HiSeq sequencer developed by Illumina. This synthesizer can generate 60 bp oligonucleotides in a pure form for immediate use within 6 h.
As there is moderate progress in increasing elongation cycle efficiencies, the use of microarray technologies to produce multiple sequences in parallel is being developed. Although further investment may be required to develop these technologies, they will improve DNA synthesis. Although new, more effective technologies can be expected to boost and dominate the market, the emergence of one winning technology that will be pursued by one vendor is unlikely. Ultimately, the development of several similarly effective technologies will ensure that DNA is priced similarly by all vendors, for example, per gene or length, making the supply of DNA of unlimited lengths affordable for the end user. Every technology is a matter of specialist developments but must eventually subject to automation, reducing the dependence of the end user on the expert involvement and deskilling DNA synthesis (Fig. 7 ).
Barriers to entry for customers
Custom DNA synthesis remains an expensive endeavour (for example, US$300–1,000 per 3 kb gene or $0.1–0.3 b −1 ). Prices vary depending on vendor, sequence composition and length. A general trend is observed towards the decrease of price to $0.01 b −1 for gene synthesis over several years 15 , for example, the current price offered by Twist Bioscience is $0.07 b −1 for gene fragments. More significant funding is required to aid research aiming to make large DNA. More specialized equipment is required for the end users to make DNA that is more complex than plasmids. The provision of such complex and large DNA can be outsourced to DNA synthesis providers (for example, Ribbon Biolabs for assembly). The complexity of custom DNA made for a particular application defines the skill barrier required for the synthesis. There are general trends for reducing the dependence on expert involvement by reducing the need to troubleshoot the DNA synthesis, which is achieved by advances in the performance of enzymes and DNA assembly methods.
Improved access to DNA in bulk quantities and enhanced information capacity of genome-sized DNA may promote further demand. Therefore, the limitation on the lengths available remains the main area of improvement to scale up. The demand for large DNA is anticipated to increase once the length limit of sequences has been overcome. Approaches exploiting automated on-chip gene assembly are promising solutions. Longer DNA will be more costly to produce. However, it is reasonable to expect that with more technologies able to break the size limit and more companies able to supply large DNA, the prices for synthetic DNA will be driven down. DNA storage applications may provide exceptions as these require substantial amounts of starting materials (g kg −1 ) to produce larger DNA than required for a biological application. The quantity and length of DNA needed are related to the amount of information to be stored 184 .
Laboratory requirements
An increasing range of benchtop printers will make in-house DNA synthesis viable: Cytiva’s ÄKTA Oligopilot provides up to 8 oligonucleotides per 3–4-h run, Kilobaser affords 2 oligonucleotides per 2-h run and Syntax, the first enzymatic printer, produces 96 oligonucleotides in parallel within 6 h. DNA ordered via service providers and manipulated in house may be assembled in small volume reactions, helping to scale down experimentation via miniaturization. Further benefits to reduce costs and lead times for DNA constructs have arisen from automation, miniaturization and parallelization of assembly methods, whereas the accurate sequence verification of assembled DNA benefits from the efficacy of NGS. However, a conservative estimate for an entry-level DNA synthesis laboratory starts at $200,000, which may increase depending on the length and production scale of the desired DNA.
In this regard, biofoundries provide a complementary infrastructure support for the end-users. Built on strong high-throughput handling and analysis technology platforms, these facilities may establish rapid in-house production pipelines for long double-stranded DNA and diverse variant libraries. Biofoundries typically host a version of a SynBio stack — an ecosystem of technologies, which allows to tackle a complex task by breaking it down to smaller tasks, providing the context and purpose of these tasks in the automated workflows to manipulate, assemble, analyse and organize DNA in small volumes and high throughput 185 .
DNA storage and accessibility
Starting with the first book written in DNA 186 , there has been steady interest in applying DNA to store and preserve data generated across different sectors of the society 186 , 187 , 188 , 189 , 190 , 191 , 192 . The concept of permanent, compact and low energy data storage in DNA gains traction, notably through the DNA Data Storage Alliance — an industry association which seeks to create a data storage ecosystem using DNA as a medium 190 . The data of interest are encoded in the four-letter alphabet of DNA (that is, A, C, G, T), whereas a set of high-fidelity enzymes is used to create copies of these data and an accurate sequencing technology is used to retrieve it. For example, Catalogue Technologies and Cambridge Consultants built a DNA synthesizer, which was able to encode 16 GB of Wikipedia data 189 . The ability to design encoding rules for data storage at whim may offer an elegant way to accommodate error rates or avoid specific sequence motifs that might be difficult for a given synthesis or sequencing technology. For instance, the recent expansion of the genetic alphabet by Hachimoji bases 193 creates the prospect of encoding data with eight instead of four letters of the DNA alphabet. This allows for an exponential rise in data density, with the help of engineered enzymes to incorporate, copy and read bases of such an expanded genetic code. These developments mean that DNA data storage products are possible and might rival biological applications as the main use of DNA synthesis technologies.
Oversight and standardization
The relatively new ability to make long pieces of DNA may prove impactful in genetic manipulation and control over living systems, which requires oversight and regulation worldwide. Regulatory policies have been developed in related areas; for example, through the Asilomar Conference on Recombinant DNA research, where communities have introduced self-regulatory processes for biosafety regulations 194 . Ultimately, the ability to write DNA will become accessible to non-experts. Therefore, there is a growing recognition that oversight policies are needed to mitigate the biosecurity risks of misusing DNA technologies. The introduction of new policies helps adapt existing mechanisms to assess these and evolving risks 195 . For example, an International Gene Synthesis Consortium was formed by industry to develop a common protocol to screen synthetic sequences, as well as the customers who order these sequences, thus self-regulating sequence identity 196 . However, DNA technology has advanced more rapidly than the ability to understand, monitor and regulate the risks. This is similar to CRISPR gene editing, in which the ability to engineer genomes was taken up by the end users before regulators had understood the consequences of misusing the technology for human genome editing 197 .
Natural DNA can become self-sustaining when incorporated into organisms which can subsequently embed into ecosystems permanently, raising issues around horizontal gene transfer as a key part of evolution 198 . Therefore, there is a need for a considered response and oversight of ethical factors, regulation and other risks associated with DNA synthesis. Similarly, maximizing the reproducibility and reliability of DNA synthesis is vital. Of particular importance is meeting emerging regulations which require industry to demonstrate the traceability of their products and technologies. This increasing focus on reproducibility and traceability prioritizes the need for standardization. The lack of reference materials and methods against which the performance and quality of synthetic DNA and synthesis methods can be evaluated including novel chemistries such as XNA is a key challenge. Reference materials can include DNA sequences, individual or libraries, which are traceable to the International System of Units (Système International d’Unités — the SI). Reference methods may provide synthesis procedures to benchmark the performance of commercial methods, for example, in relation to elongation cycle efficiency. Encouragingly, reference materials have become available, such as the first ‘human genome’ DNA reference material (RM 8398) from the National Institute of Standards and Technology, which can evaluate the accuracy of NGS assays 199 . New metrology, which will provide the basis of comparison and reproducibility for DNA synthesis, is required to support existing and emerging DNA synthesis technologies.
Conclusions
With the rapid development of technologies able to read DNA, the ability to write DNA has lagged behind. DNA synthesis technologies developed to date may differ in their ability to bridge the DNA writing gap (Table 1 and Fig. 1 ). However, their continuous development is driven by two main factors: the lack of methods to routinely make DNA of unlimited lengths at scale and cost and an increasing demand for DNA from different and unrelated sectors. Alongside the success of NGS, these two factors stimulate the search for innovative technologies and guarantee commercialization success for any strategy able to overcome the barrier of size-limited DNA synthesis. As a consequence, DNA makers tightly guard their knowledge and are cautious in their claims of what their technologies can deliver. This is notable given that most of the existing methodologies are similar, using the same starting materials, suggesting that innovation progresses at a marginal pace. Conversely, close competition prompts companies to search for an application niche at early stages or demonstrate the use of their technologies in producing challenging DNA molecules.
Advances in automation will make DNA synthesis increasingly more accessible to non-experts. Most vendors, especially those who provide synthetic DNA as a service, appreciate the need for oversight and regulatory policies to protect their commercial and reputational interests and may, in turn, contribute to the development of such policies. Once DNA synthesis is affordable for small hackerspaces of enthusiasts collaborating on making new DNA molecules, the uses of the produced DNA will be difficult to contain. Risk governance designed to monitor the use and distribution of synthetic DNA in accordance with applicable policies and ethics will reduce the likelihood of adverse events.
To conclude, new synthesis methods will continue to emerge with a persistent focus on providing greener solutions, mitigating potentially harmful consequences for the environment owing to the use of organic solvents and hazardous chemicals. With the limitations of existing synthetic approaches, it is unlikely that a routine methodology to effectively synthesize size-unlimited DNA will soon be available. Yet, there remains plenty of space in the gene writing gap for breakthroughs in the foreseeable future.
Change history
30 june 2023.
A Correction to this paper has been published: https://doi.org/10.1038/s41570-023-00521-x
Hughes, R. A. & Ellington, A. D. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol. 9 , a023812 (2017).
Article PubMed PubMed Central Google Scholar
Kosuri, S. & Church, G. M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11 , 499–507 (2014).
Article CAS PubMed PubMed Central Google Scholar
Rothemund, P. W. K. Folding DNA to create nanoscale shapes and patterns. Nature 440 , 297–302 (2006).
Article CAS PubMed Google Scholar
Pinheiro, V. B. & Holliger, P. Towards XNA nanotechnology: new materials from synthetic genetic polymers. Trends Biotechnol. 32 , 321–328 (2014).
Lee, I. & Berdis, A. J. Non-natural nucleotides as probes for the mechanism and fidelity of DNA polymerases. BBA Proteins Proteom. 1804 , 1064–1080 (2010).
Article CAS Google Scholar
Douglas, S. M., Bachelet, I. & Church, G. M. A logic-gated nanorobot for targeted transport of molecular payloads. Science 335 , 831–834 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339 , 819–823 (2013).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17 , 333–351 (2016).
Schatz, M. C. & Phillippy, A. M. The rise of a digital immune system. Gigascience 1 , 14 (2012).
Article Google Scholar
McLennan, A. Regulation of Synthetic Biology (Edward Elgar, 2018).
Schwab, K. The fourth industrial revolution: what it means and how to respond. Foreign Affairs https://www.foreignaffairs.com/world/fourth-industrial-revolution (2015).
Leurent, H. & Abbosh, O. Shaping the sustainability of production systems: fourth industrial revolution technologies for competitiveness and sustainable growth (World Economic Forum, 2019).
Chui, M., Evers, M., Manyika, J., Zheng, A. & Nisbet, T. The Bio Revolution (McKinsey Global Institute, 2020).
Collins, F. S. et al. Finishing the euchromatic sequence of the human genome. Nature 431 , 931–945 (2004).
Carlson, R. in Synthetic Biology: Parts, Devices and Applications Ch. 1 (ed. Smolke, C.) 3–13 (Wiley-Blackwell, 2018). The first detailed analysis of gene sequencing and gene synthesis technologies in light of Moore’s law highlighting the gap between our ability to read and write DNA .
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8 , 2281–2308 (2013).
Blakney, A. K. & Bekker, L. G. DNA vaccines join the fight against COVID-19. Lancet 399 , 1281–1282 (2022).
Han, S. H., Park, Y. J. & Park, C. M. HOS1 activates DNA repair systems to enhance plant thermotolerance. Nat. Plants 6 , 1439–1446 (2020).
Brophy, J. A. N. et al. Synthetic genetic circuits as a means of reprogramming plant roots. Science 377 , 747–751 (2022).
Watson, J. D. & Crick, F. H. C. Molecular structure of nucleic acids: a structure for deoxyribose nucelic acid. Nature 171 , 737–738 (1953).
Cumbers, J. A New Way of Making DNA is About to Revolutionize the Biotech Industry (Forbes, 2020).
Reisch, M. S. Danaher buys oligonucleotide maker Integrated DNA Technologies. Chem. Eng. News 96 , 10–10 (2018).
Google Scholar
Eisenstein, M. Enzymatic DNA synthesis enters new phase. Nat. Biotechnol. 38 , 1113–1115 (2020).
Mohsen, M. G. & Kool, E. T. The discovery of rolling circle amplification and rolling circle transcription. Acc. Chem. Res. 49 , 2540–2550 (2016).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6 , 343–345 (2009). The seminal paper introducing a revolutionary method enabling the assembly of multiple DNA fragments in a single reaction — Gibson assembly .
Stemmer, W. P., Crameri, A., Ha, K. D., Brennan, T. M. & Heyneker, H. L. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164 , 49–53 (1995).
Jensen, M. A. & Davis, R. W. Template-independent enzymatic oligonucleotide synthesis (TiEOS): its history, prospects, and challenges. Biochemistry 57 , 1821–1832 (2018).
Sarac, I. & Hollenstein, M. Terminal deoxynucleotidyl transferase in the synthesis and modification of nucleic acids. ChemBioChem 20 , 860–871 (2018).
Crosby, S. et al. Oligonucleotide and nucleic acid synthesis. Patent WO/2019/145713 (2019).
Kosuri, S. et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat. Biotechnol. 28 , 1295–1299 (2010).
Kodumal, S. J. et al. Total synthesis of long DNA sequences: synthesis of a contiguous 32-kb polyketide synthase gene cluster. Proc. Natl Acad. Sci. USA 101 , 15573–15578 (2004).
Michelson, A. M. & Todd, A. R. Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage. J. Chem. Soc. 1 , 2632–2638 (1955).
Nishimura, S., Jones, D. S. & Khorana, H. G. Studies on polynucleotides. 48. The in vitro synthesis of a co-polypeptide containing two amino acids in alternating sequence dependent upon a DNA-like polymer containing two nucleotides in alternating sequence. J. Mol. Biol. 13 , 302–324 (1965).
Merrifield, R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc . 85 , 2149–2154 (1963).
Letsinger, R. L. & Mahadevan, V. Oligonucleotide synthesis on a polymer support. J. Am. Chem. Soc. 87 , 3526–3527 (1965). Inception paper for chemical oligonucleotide synthesis .
Beaucage, S. L. & Caruthers, M. H. Deoxynucleoside phosphoramidites — a new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 22 , 1859–1862 (1981). Introduction of phosphoramidite method for oligonucleotide synthesis that continues to underpin DNA synthesis today .
Matteucci, M. D. & Caruthers, M. H. Synthesis of deoxyoligonucleotides on a polymer support. J. Am. Chem. Soc. 103 , 3185–3191 (1981).
McBride, L. J. & Caruthers, M. H. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 24 , 245–248 (1983).
Vinayak, R. Chemical synthesis, analysis, and purification of oligoribonucleotides. Methods 5 , 7–18 (1993).
Caruthers, M. H. et al. Chemical synthesis of deoxynucleotides by the phosphoramidite method. Method. Enzymol. 154 , 287–313 (1987).
Letsinger, R. L., Caruthers, M. H. & Jerina, D. M. Reactions of nucleosides on polymer supports. Synthesis of thymidylylthymidylylthymidine. Biochemistry 6 , 1379–1388 (1967).
Caruthers, M. H. The chemical synthesis of DNA/RNA: our gift to science. J. Biol. Chem. 288 , 1420–1427 (2013).
Caruthers, M. H. & Beaucage, S. L. Phosphoramidite compounds and processes. US Patent 4,415,732 (1983).
Caruthers, M. H. & Matteucci, M. D. Nucleosides useful in the preparation of polynucleotides. US Patent 4,500,707 (1985).
Caruthers, M. H. & Beaucage, S. L. Phosphoramidite nucleoside compounds. US Patent 4,668,777 (1987).
Pon, R. T. Solid‐phase supports for oligonucleotide synthesis. Curr. Protoc. Nucleic Acid Chem. 3 , 1–28 (2000).
LeProust, E. M. et al. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res. 38 , 2522–2540 (2010). High impact methodology for generating libraries of small DNA sequences while avoiding depurination during synthesis .
Hughes, T. R. et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 19 , 342–347 (2001).
LeProust, E., Zhang, H., Yu, P., Zhou, X. & Gao, X. Characterization of oligodeoxyribonucleotide synthesis on glass plates. Nucleic Acids Res . 29 , 2171–2180 (2001).
Cleary, M. A. et al. Production of complex nucleic acid libraries using highly parallel in situ oligonucleotide synthesis. Nat. Methods 1 , 241–248 (2004).
Ferguson, A. J. et al. Thermofluidic chip containing virtual thermal wells. Eng. Biol. 3 , 20–23 (2019).
Crosby, S. Unlocking synthetic biology through DNA synthesis. Chim. Oggi 38 , 22–24 (2020).
CAS Google Scholar
Letsinger, R. L. & Ogilvie, K. K. Nucleotide chemistry. XIII. Synthesis of oligothymidylates via phosphotriester intermediates. J. Am. Chem. Soc. 91 , 3350–3355 (1969).
Letsinger, R. L., Finnan, J. L., Heavner, G. A. & Lunsford, W. B. Nucleotide chemistry. XX. Phosphite coupling procedure for generating internucleotide links. J. Am. Chem. Soc . 97 , 3278–3279 (1975).
Letsinger, R. L. & Lunsford, W. B. Synthesis of thymidine oligonucleotides by phosphite triester intermediates. J. Am. Chem. Soc. 98 , 3655–3661 (1976).
Smith, M., Rammler, D. H., Goldberg, I. H. & Khorana, H. G. Studies on polynucleotides. XIV. Specific synthesis of the C3″-C5″ interribonucleotide linkage. Syntheses of uridylyl-(3″→5″)uridine and uridylyl-(3″→5″)-adenosine. J. Am. Chem. Soc. 84 , 430–440 (1962).
Schaller, H., Weimann, G., Lerch, B. & Khorana, H. G. Studies on polynucleotides. XXIV.1 The stepwise synthesis of specific deoxyribopolynucleotides. Protected derivatives of deoxyribonucleosides and new syntheses of deoxyribonucleoside-3″ phosphates. J. Am. Chem. Soc. 85 , 3821–3827 (1963).
Krotz, A. H. et al. Solution stability and degradation pathway of deoxyribonucleoside phosphoramidites in acetonitrile. Nucleosides Nucleotides Nucleic Acids 23 , 767–775 (2004).
Caruthers, M. & Matteucci, M. D. Process for preparing polynucleotides. US Patent 4,458,066 (1984).
Caruthers, M. H. & Beaucage, S. L. Process for oligonucleotide synthesis using phosphoramidite intermediates. US Patent 4,973,679 (1990).
Koster, H. & Sinha, N. D. Process for the preparation of oligonucleotides. US Patent 4,725,677 (1988).
Efcavitch, J. W. & Heiner, C. Depurination as a yield decreasing mechanism in oligodeoxynucleotide synthesis. Nucleosides Nucleotides 4 , 267 (1985).
An, R. et al. Non-enzymatic depurination of nucleic acids: factors and mechanisms. PLoS ONE 9 , e0115950 (2014).
Suzuki, T., Ohsumi, S. & Makino, K. Mechanistic studies on depurination and apurinic site chain breakage in oligodeoxyribonucleotides. Nucleic Acids Res. 22 , 4997–5003 (1994).
Ravikumar, V., Andrade, M., Mulvey, D. & Cole., D. L. Carbocation scavenging during oligonucleotide synthesis. US Patent 5,510,476A (1996).
Wooddell, C. I. & Burgess, R. R. Use of asymmetric PCR to generate long primers and single-stranded DNA for incorporating cross-linking analogs into specific sites in a DNA probe. Genome Res. 6 , 886–892 (1996).
Veneziano, R. et al. Designer nanoscale DNA assemblies programmed from the top down. Science 352 , e1534 (2016).
Veneziano, R. et al. In vitro synthesis of gene-length single-stranded DNA. Sci. Rep. 8 , e6548 (2018).
Mitchell, L. A. et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 355 , e1045 (2017).
Reddy, M. P., Hanna, N. B. & Farooqui, F. Ultrafast cleavage and deprotection of oligonucleotides synthesis and use of C Ac derivatives. Nucleosides Nucleotides 16 , 1589–1598 (1997).
Delarue, M. et al. Crystal structures of a template-independent DNA polymerase: murine terminal deoxynucleotidyltransferase. EMBO J. 21 , 427–439 (2002).
Bollum, F. J. Chemically defined templates and initiators for deoxypolynucleotide synthesis. Science 144 , 560–560 (1964).
Boulé, J. B., Rougeon, F. & Papanicolaou, C. Terminal deoxynucleotidyl transferase indiscriminately incorporates ribonucleotides and deoxyribonucleotides. J. Biol. Chem. 276 , 31388–31393 (2001).
Article PubMed Google Scholar
Lee, H. H., Church, G. M. & Kalhor, R. Enzymatic nucleic acid synthesis. Patent WO/2017/176541 (2017).
Ybert, T. & Gariel, S. Method for synthesizing nucleic acids, in particular long nucleic acids, use of said method and kit for implementing said method. US Patent 2021/0130863 (2021).
Ybert, T. & Gariel, S. Modified nucleotides for synthesis of nucleic acids, a kit containing such nucleotides and their use for the production of synthetic nucleic acid sequences or genes. US Patent 2020/0231619 (2020).
Creton, S. Efficient product cleavage in template-free enzymatic synthesis of polynucleotides. Patent WO/2020/165137 (2020).
Loftie-Eaton, W. et al. Novel variants of endonuclease V and uses thereof. Patent WO/2022/090057 (2022).
Schott, H. & Schrade, H. Single‐step elongation of oligodeoxynucleotides using terminal deoxynucleotidyl transferase. Eur. J. Biochem. 143 , 613–620 (1984).
Motea, E. A. & Berdis, A. J. Terminal deoxynucleotidyl transferase: the story of a misguided DNA polymerase. Biochim. Biophys. Acta 1804 , 1151–1166 (2010).
Efcavitch, J. W. & Sylvester, J. E. Modified template-independent enzymes for polydeoxynucleotide synthesis. US Patent 2016/0108382 (2016). Invention exacting an effective reversible termination mechanism with NTPs modified with terminators to add a single, defined nucleotide per reaction step .
Hiatt, A. C. & Rose, F. D. Enzyme catalyzed template-independent creation of phosphodiester bonds using protected nucleotides. US Patent 5,990,300 (1999).
Barthel, S., Palluk, S., Hillson, N. J., Keasling, J. D. & Arlow, D. H. Enhancing terminal deoxynucleotidyl transferase activity on substrates with 3′ terminal structures for enzymatic de novo DNA synthesis. Genes 11 , e102 (2020).
Deibel, M. R. & Coleman, M. S. Biochemical properties of purified human terminal deoxynucleotidyltransferase. J. Biol. Chem. 255 , 4206–4212 (1980).
Chang, L. M. S. & Bollum, F. J. Multiple roles of divalent cation in the terminal deoxynucleotidyltransferase reaction. J. Biol. Chem. 265 , 17436–17440 (1990).
Chang, L. M. & Bollum, F. J. Deoxynucleotide-polymerizing enzymes of calf thymus gland. V. Homogeneous terminal deoxynucleotidyl transferase. J. Biol. Chem. 246 , 909–916 (1971).
Bhan, N. et al. Recording temporal signals with minutes resolution using enzymatic DNA synthesis. J. Am. Chem. Soc. 143 , 16630–16640 (2021).
Arzumanov, A. A., Victorova, L. S., Jasko, M. V., Yesipov, D. S. & Krayevsky, A. A. Terminal deoxynucleotidyl transferase catalyzes the reaction of DNA phosphorylation. Nucleic Acids Res. 28 , 1276–1281 (2000).
Krayevsky, A., Victorova, L. S., Arzumanov, A. A. & Jasko, M. V. Terminal deoxynucleotidyl transferase catalysis of DNA (oligodeoxynucleotide) phosphorylation. Pharmacol. Ther. 85 , 165–173 (2000).
Flamme, M. et al. Evaluation of 3′-phosphate as a transient protecting group for controlled enzymatic synthesis of DNA and XNA oligonucleotides. Commun. Chem. 5 , e68 (2022).
Collins, J., Singh, S. & Vanier, G. Microwave technology for solid phase peptide synthesis. Chim. Oggi 30 , 26–29 (2012).
Hari Das, R., Ahirwar, R., Kumar, S. & Nahar, P. Microwave-assisted rapid enzymatic synthesis of nucleic acids. Nucleosides Nucleotides Nucleic Acids 35 , 363–369 (2016).
Yoshimura, T., Sugiyama, J., Mineki, S. & Ohuchi, S. Effect of Microwaves on DNA and Proteins (Springer, 2017).
DNA Script. Enzymatic DNA synthesis: technical note (DNA Script, 2021).
Soskine, M., Champion, E. & Mchale, D. Stabilized N-terminally truncated terminal deoxynucleotidyl transferase variants and uses thereof. Patent WO/2022/063835 (2022).
Hutter, D. et al. Labeled nucleoside triphosphates with reversibly terminating aminoalkoxyl groups. Nucleosides Nucleotides Nucleic Acids 29 , 879–895 (2010).
Chen, M. C., Huang, J., Lazar, R. & McInroy, G. Azidomethyl ether deprotection method. US Patent 2018/0201968 (2018).
Hoff, K., Halpain, M., Garbagnati, G., Edwards, J. S. & Zhou, W. Enzymatic synthesis of designer DNA using cyclic reversible termination and a universal template. ACS Synth. Biol. 9 , 283–293 (2020).
Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 , 53–59 (2008).
Guo, J. et al. Four-color DNA sequencing with 3′-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proc. Natl Acad. Sci. USA 105 , 9145–9150 (2008).
Stemple, D. L., Mankowska, S. A. & Harvey, S. A. Compositions and methods for template free enzymatic nucleic acid synthesis. Patent WO/2018/152323 (2018).
Mathews, A. S., Yang, H. & Montemagno, C. Photo-cleavable nucleotides for primer free enzyme mediated DNA synthesis. Org. Biomol. Chem. 14 , 8278–8288 (2016).
Metzker, M. L. et al. Termination of DNA synthesis by novel 3′-modified deoxyribonucleoside 5′-triphosphates. Nucleic Acids Res. 22 , 4259–4267 (1994).
Wu, W. et al. Termination of DNA synthesis by N6-alkylated, not 3′-O-alkylated, photocleavable 2′-deoxyadenosine triphosphates. Nucleic Acids Res. 35 , 6339–6349 (2007).
Bowers, J. et al. Virtual terminator nucleotides for next-generation DNA sequencing. Nat. Methods 6 , 593–595 (2009).
Takeshita, L., Yamada, Y., Masaki, Y. & Seio, K. Synthesis of deoxypseudouridine 5′-triphosphate bearing the photoremovable protecting group at the N1 position capable of enzymatic incorporation to DNA. J. Org. Chem. 85 , 1861–1870 (2020).
Flamme, M. et al. Towards the enzymatic synthesis of phosphorothioate containing LNA oligonucleotides. Bioorg. Med. Chem. Lett. 48 , e128242 (2021).
Wojciechowski, F. & Ybert, T. Method for preparing 3’-O-amino-2’-deoxyribonucleoside-5’-triphosphates. Patent WO/2021/198040 (2021).
Champion, E., Soskine, M., Ybert, T. & Delarue, M. Variants of terminal deoxynucleotidyl transferase and uses thereof. Patent WO/2019/135007 (2019). Underpinning technology by DNA Script for template-independent DNA synthesis using re-engineered TdT .
Ybert, T. Template-free enzymatic polynucleotide synthesis using dismutationless terminal deoxynucleotidyl transferase variants. Patent WO/2021/122539 (2021).
Champion, E., Soskine, M., Jaziri, F. & Mchale, D. Chimeric terminal deoxynucleotidyl transferases for template-free enzymatic synthesis of polynucleotides. Patent WO/2021/122539 (2021).
Soskine, M. & Champion, E. Terminal deoxynucleotidyl transferase variants and uses thereof. Patent WO/2021/213903 (2021).
Lu, X. et al. Enzymatic DNA synthesis by engineering terminal deoxynucleotidyl transferase. ACS Catal. 12 , 2988–2997 (2022).
Chua, J. P. S. et al. Evolving a thermostable terminal deoxynucleotidyl transferase. ACS Synth. Biol. 9 , 1725–1735 (2020).
Chen, M. C. & McInroy, G. R. Method of oligonucleotide synthesis. Patent WO/2020/178604 (2020).
McInroy, G. R., Ost, T. W. B. & Lovedale, D. Methods relating to de novo enzymatic nucleic acid synthesis. Patent WO/2022/034331 (2022).
Chen, M. C., McInroy, G. R., Fox, M. E. & Matuszewski, M. R. Nucleic acid polymer with amine-masked bases. Patent WO/2020/229831 (2020).
Chen, M. C., Lazar, R. A., Huang, J. & McInroy, G. R. A process for the preparation of nucleic acid by means of 3′-o-azidomethyl nucleotide triphosphate. Patent WO/2016/139477 (2016).
Ost, T. W. B., McInroy, G. R., Gaber, Z. B., Swerdlow, H. & Tognoloni, C. Methods of nucleic acid synthesis. Patent WO/2021/148809 (2021).
Champion, E. et al. High efficiency template-free enzymatic synthesis of polynucleotides. Patent WO/2021/094251 (2021).
Palluk, S. et al. De novo DNA synthesis using polymerase–nucleotide conjugates. Nat. Biotechnol. 36 , 645–650 (2018).
Chang, L. M. S., Bollum, F. J. & Gallo, R. C. Molecular biology of terminal transferase. Crit. Rev. Biochem. 21 , 27–52 (1986).
Knapp, D. C. et al. Fluoride-cleavable, fluorescently labelled reversible terminators: synthesis and use in primer extension. Chem. Eur. J. 17 , 2903–2915 (2011).
Chen, M. C. & McInroy, G. R. Nucleotide derivatives containing amine masked moieties and their use in a templated and non-templated enzymatic nucleic acid synthesis. Patent WO/2019/097233 (2019).
Chen, M. C., Lazar, R. A., Huang, J. & McInroy, G. R. Compositions and methods related to nucleic acid synthesis. US Patent 11236377 (2020).
McInroy, G. R., Cook, I. H., Chen, M. C. & Chen, S. Modified terminal deoxynucleotidyl transferase (Tdt) enzymes. Patent WO/2022/029427 (2022).
Chen, M. C., Huang, J. & McInroy, G. R. Use of terminal transferase enzyme in nucleic acid synthesis. Patent WO/2018/215803 (2018).
Bell, N. M., Mankowska, S. A., Harvey, S. A. & Stemple, D. L. gSynth: synthesis and assembly of whole plasmids (Camena Bioscience, 2020).
Efcavitch, W. & Siddiqi, S. Methods and apparatus for synthesizing nucleic acids. US Patent 8,808,989 (2014). Invention for template-free de novo DNA synthesis, nucleotide by nucleotide, by Molecular Assemblies .
Aggarwal, S. Reusable initiators for synthesizing nucleic acids. Patent WO/2021/207158 (2021).
Efcavitch, J. W. & Tubbs, J. L. Nucleic acid synthesis using DNA polymerase theta. Patent WO/2018/175436 (2018).
Arlow, D. H. & Palluk, S. Nucleic acid synthesis and sequencing using tethered nucleoside triphosphates. US Patent 2019/0112627 (2019). Invention of exemplar reversible termination by ANSA Biotechnologies to sterically control prescribed polymerization using TdT-tethered NTPs .
Bell, N. M., Mankowska, S. A., Harvey, S. A., Stemple, D. L. & Fraser, A. gSynth: a highly accurate, enzymatic, de novo synthesis and gene assembly technology (Camena Bioscience, 2019).
Stemple, D. L., Fraser, A. G., Mankowska, S. & Bell, N. Compositions and methods for template-free geometric enzymatic nucleic acid synthesis. Patent WO/2020/150143 (2020). Exemplar methodology by Camena Bioscience for template-free nucleic acid synthesis using reversible terminating NTPs .
Horgan, A., Sarvac, I., Niyomchon, S. & Godron, X. Increasing long-sequence yields in template-free enzymatic synthesis of polynucleotides. Patent WO/2021/018921 (2021).
Hutchison, C. A. III et al. Design and synthesis of a minimal bacterial genome. Science 351 , e6253 (2016).
Casini, A., Storch, M., Baldwin, G. S. & Ellis, T. Bricks and blueprints: methods and standards for DNA assembly. Nat. Rev. Mol. Cell Biol. 16 , 568–576 (2015).
Young, R., Haines, M., Storch, M. & Freemont, P. S. Combinatorial metabolic pathway assembly approaches and toolkits for modular assembly. Metab. Eng. 63 , 81–101 (2021).
Gibson, D. G. et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319 , 1215–1220 (2008).
Gibson, D. G., Smith, H. O., Hutchison, C. A. III, Venter, J. C. & Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat. Methods 7 , 901–903 (2010).
TerMaat, J. R., Pienaar, E., Whitney, S. E., Mamedov, T. G. & Subramanian, A. Gene synthesis by integrated polymerase chain assembly and PCR amplification using a high-speed thermocycler. J. Microbiol. Methods 79 , 295–300 (2009).
Roth, T. L., Milenkovic, L. & Scott, M. P. A rapid and simple method for DNA engineering using cycled ligation assembly. PLoS ONE 9 , e107329 (2014).
Sayers, J. R. & Eckstein, F. Properties of overexpressed phage T5 D15 exonuclease. Similarities with Escherichia coli DNA polymerase I 5′-3′ exonuclease. J. Biol. Chem. 265 , 18311–18317 (1990).
Housby, J. N., Thorbjarnardóttir, S. H., Jónsson, Z. O. & Southern, E. M. Optimised ligation of oligonucleotides by thermal ligases: comparison of Thermus scotoductus and Rhodothermus marinus DNA ligases to other thermophilic ligases. Nucleic Acids Res. 28 , e10 (2000).
Takahashi, M., Yamaguchi, E. & Uchida, T. Thermophilic DNA ligase. Purification and properties of the enzyme from Thermus thermophilus HB8. J. Biol. Chem. 259 , 10041–10047 (1984).
Saiki, R. K. et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239 , 487–491 (1988).
Jayaraman, K., Fingar, S. A., Shah, J. & Fyles, J. Polymerase chain reaction-mediated gene synthesis: synthesis of a gene coding for isozyme c of horseradish peroxidase. Proc. Natl Acad. Sci. USA 88 , 4084–4088 (1991).
Saiki, R. K. et al. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anaemia. Science 230 , 1350–1354 (1985).
Hayes, M. J., Ferguson, A. J., Juncu, V. D. & Temple, S. Temperature control device. Patent WO/2018/104698 (2018).
Hayes, M. J., Sanches-Kuiper, R. M. & Bygrave, D. A. Error detection during hybridization of target double-stranded nucleic acid. Patent WO/2019/064006 (2019).
Crosby, S. R., Jennison, M. & Brennan, J. Thermally-cleavable protecting and linker groups. Patent WO/2018/189546 (2018).
Brennan, J., Bygrave, D., Aditya, S. & Sanches-Kuiper, R. Method for producing double stranded polynucleotides based on oligonucleotides with selected and different melting temperatures. Patent WO/2018/167475 (2018). Enabling methodology of oligonucleotide selectivity for highly parallelized DNA synthesis under thermal control developed by Evonetix .
Vladar, H. P., Redondo, F. & Aparecido, R. A novel method for synthesis of polynucleotides using a diverse library of oligonucleotides. Patent WO/2019/073072 (2019). Invention of a highly selective methodology by Ribbon Biolabs for DNA synthesis from diverse oligonucleotide libraries culminating in the first synthesis of >10 kb DNA .
Vladar, H. P., Redondo, F. & Aparecido, R. A novel method for synthesis of polynucleotides using a diverse library of oligonucleotides. US Patent 2020/0283756 (2020).
Rouillard, J. M. et al. Gene2Oligo: oligonucleotide design for in vitro gene synthesis. Nucleic Acids Res. 32 , W176–W180 (2004).
Banyai, W., Chen, S., Fernandez, A., Indermuhle, P. & Peck, B. J. De novo synthesized gene libraries. Patent WO/2015/021080 (2015).
Peck, B. J., Noe, M., Pitsch, S. & Weiss, P. A. Highly accurate de novo polynucleotide synthesis. Patent WO/2020/139871 (2020).
Horgan, A., Lachaize, H., Verado, D. & Godron, X. Massively parallel enzymatic synthesis of polynucleotides. Patent WO/2022/013094 (2022).
Gao, X. et al. Oligonucleotide synthesis using solution photogenerated acids. J. Am. Chem. Soc. 120 , 12698–12699 (1998).
Gao, X. et al. A flexible light-directed DNA chip synthesis gated by deprotection using solution photogenerated acids. Nucleic Acids Res. 29 , 4744–4750 (2001).
Fodor, S. P. A. et al. Light-directed, spatially addressable parallel chemical synthesis. Science 251 , 767–773 (1991).
Zhou, X. et al. Microfluidic PicoArray synthesis of oligodeoxynucleotides and simultaneous assembling of multiple DNA sequences. Nucleic Acids Res. 32 , 5409–5417 (2004).
Septak, M. Kinetic studies on depurination and detritylation of CPG-bound intermediates during oligonucleotide synthesis. Nucleic Acids Res. 24 , 3053–3058 (1996).
Warner, J. R., Reeder, P. J., Karimpour-Fard, A., Woodruff, L. B. A. & Gill, R. T. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat. Biotechnol. 28 , 856–862 (2010).
Tian, J. et al. Accurate multiplex gene synthesis from programmable DNA microchips. Nature 432 , 1050–1054 (2004).
Engler, C., Kandzia, R. & Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 3 , e3647 (2008).
Nugent, R., Chen, S., Kettleborough, R. & Raynard, N. Barcode-based nucleic acid sequence assembly. Patent WO/2020/257612 (2020).
Smith, H. O., Hutchison, C. A. III, Pfannkoch, C. & Venter, J. C. Generating a synthetic genome by whole genome assembly: φX174 bacteriophage from synthetic oligonucleotides. Proc. Natl Acad. Sci. USA 100 , 15440–15445 (2003).
Schaudy, E., Lietard, J. & Somoza, M. M. Sequence preference and initiator promiscuity for de novo DNA synthesis by terminal deoxynucleotidyl transferase. ACS Synth. Biol. 10 , 1750–1760 (2021).
Chow, D. C., Lee, W. K., Zauscher, S. & Chilkoti, A. Enzymatic fabrication of DNA nanostructures: extension of a self-assembled oligonucleotide monolayer on gold arrays. J. Am. Chem. Soc. 127 , 14122–14123 (2005).
Lee, H. et al. Photon-directed multiplexed enzymatic DNA synthesis for molecular digital data storage. Nat. Commun. 11 , e5246 (2020).
Deshpande, S., Yang, Y., Chilkoti, A. & Zauscher, S. Enzymatic synthesis and modification of high molecular weight DNA using terminal deoxynucleotidyl transferase. Method. Enzymol. 627 , 163–188 (2019).
Scott, V. L. et al. Novel synthetic plasmid and Doggybone TM DNA vaccines induce neutralizing antibodies and provide protection from lethal influenza challenge in mice. Hum. Vacc. Immunother. 11 , 1972–1982 (2015).
Walters, A. A. et al. Comparative analysis of enzymatically produced novel linear DNA constructs with plasmids for use as DNA vaccines. Gene Ther. 21 , 645–652 (2014).
Banér, J., Nilsson, M., Mendel-Hartvig, M. & Landegren, U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 26 , 5073–5078 (1998).
Hill, V. Production of closed linear DNA. Patent WO/2010/086626 (2010). Enzymatic methodology for manufacturing dbDNA as the first plasmid-free vector cassette encoding the sequence of interest invented by Touchlight Genetics .
Kendirgi, F. et al. Novel linear DNA vaccines induce protective immune responses against lethal infection with influenza virus type A/H5N1. Hum. Vaccines 4 , 410–419 (2008).
Karbowniczek, K. et al. Doggybone TM DNA: an advanced platform for AAV production. Cell Gene Ther. Insights 3 , 731–738 (2017).
Huang, W. M., Joss, L., Hsieh, T. & Casjens, S. Protelomerase uses a topoisomerase IB/Y-recombinase type mechanism to generate DNA hairpin ends. J. Mol. Biol. 337 , 77–92 (2004).
Smith, H. O. & Wilcox, K. W. A restriction enzyme from hemophilus influenzae. J. Mol. Biol. 51 , 389–391 (1970).
Porter, N., Rothwell, P. J., Gehrig-Giannini, S., Babula, A. & Adie, T. A. J. Synthesis of DNA with improved yield. Patent WO/2020/035698 (2020).
Porter, N., Rothwell, P. & Extance, J. Synthesis of DNA. Patent WO/2016/034849 (2016).
Porter, N. Production of closed linear DNA using a palindromic sequence. Patent WO/2012/017210 (2012).
Extance, A. How DNA could store all the world’s data. Nature 537 , 22–24 (2016).
Hillson, N. et al. Building a global alliance of biofoundries. Nat. Commun. 10 , e2040 (2019).
Church, G., Gao, Y. & Kosuri, S. Next-generation digital information storage in DNA. Science 337 , e1226355 (2012). The first book written using DNA demonstrating DNA as a digital medium for information storage .
Lee, H. H., Kalhor, R., Goela, N., Bolot, J. & Church, G. M. Terminator-free template-independent enzymatic DNA synthesis for digital information storage. Nat. Commun. 10 , e2383 (2019).
Ceze, L., Nivala, J. & Strauss, K. Molecular digital data storage using DNA. Nat. Rev. Genet. 20 , 456–466 (2019).
Vitak, S. Technology alliance boosts efforts to store data in DNA. Nature https://doi.org/10.1038/d41586-021-00534-w (2021).
Xu, C., Zhao, C., Ma, B. & Liu, H. Uncertainties in synthetic DNA-based data storage. Nucleic Acids Res. 49 , 5451–5469 (2021).
Song, L. F., Deng, Z. H., Gong, Z. Y., Li, L. L. & Li, B. Z. Large-scale de novo oligonucleotide synthesis for whole-genome synthesis and data storage: challenges and opportunities. Front. Bioeng. Biotechnol. 9 , e689797 (2021).
Meiser, L. C. et al. Reading and writing digital data in DNA. Nat. Protoc. 15 , 86–101 (2020).
Hoshika, S. et al. Hachimoji DNA and RNA: a genetic system with eight building blocks. Science 363 , 884–887 (2020).
Berg, P. Asilomar 1975: DNA modification secured. Nature 455 , 290–291 (2008).
Fatehi, L. & Hall, R. F. Synthetic biology in the FDA realm: toward productive oversight assessment. Food Drug Law J. 70 , 339–369 (2015).
PubMed Google Scholar
International Gene Synthesis Consortium. About IGSC. International Gene Synthesis Consortium https://genesynthesisconsortium.org .
National Academy of Medicine, National Academy of Sciences & the Royal Society. Heritable human genome editing (National Academies, 2020).
Burmeister, A. R. Horizontal gene transfer. Evol. Med. Public Health 1 , 193–194 (2015).
Kalman, L. V. et al. Development and characterization of reference materials for genetic testing: focus on public partnerships. Ann. Lab. Med. 36 , 513–520 (2016).
Spencer, M. The stereochemistry of deoxyribonucleic acid. II. Hydrogen-bonded pairs of bases. Acta Crystallogr. 12 , 66–71 (1959).
Download references
Acknowledgements
The authors acknowledge the UK Defence Science and Technology Laboratory for initiating and supporting this review. The authors thank P. Oyston for the critical assessment of the work.
Author information
Authors and affiliations.
National Physical Laboratory, Teddington, Middlesex, UK
Alex Hoose & Maxim G. Ryadnov
BiologIC Technologies, Cambridge, UK
Richard Vellacott
London Biofoundry, Translation and Innovation Hub, Imperial College White City Campus, London, UK
Marko Storch & Paul S. Freemont
Section of Structural and Synthetic Biology, Faculty of Medicine, Imperial College London, London, UK
You can also search for this author in PubMed Google Scholar
Contributions
All authors researched data for the article. All authors contributed substantially to discussion of the content. A.H. and M.G.R. wrote the article. All authors reviewed and edited the manuscript before submission.
Corresponding author
Correspondence to Maxim G. Ryadnov .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Peer review
Peer review information.
Nature Reviews Chemistry thanks Marcel Hollenstein, De-Ming Kong, Vincent Noireaux and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Hoose, A., Vellacott, R., Storch, M. et al. DNA synthesis technologies to close the gene writing gap. Nat Rev Chem 7 , 144–161 (2023). https://doi.org/10.1038/s41570-022-00456-9
Download citation
Accepted : 29 November 2022
Published : 23 January 2023
Issue Date : March 2023
DOI : https://doi.org/10.1038/s41570-022-00456-9
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Dna as a universal chemical substrate for computing and data storage.
- Bas W. A. Bögels
- Tom F. A. de Greef
Nature Reviews Chemistry (2024)
Recent progress in DNA data storage based on high-throughput DNA synthesis
- Haewon Shin
- Honggu Chun
Biomedical Engineering Letters (2024)
Recent Progress in High-Throughput Enzymatic DNA Synthesis for Data Storage
- Sung-Yune Joe
BioChip Journal (2024)
Fast and efficient template-mediated synthesis of genetic variants
- Yiming Huang
- Harris H. Wang
Nature Methods (2023)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

The current status and trends of DNA extraction
Affiliations.
- 1 Department of Chemistry, Faculty of Science, Hong Kong Baptist University (HKBU), Hong Kong, Hong Kong.
- 2 Division of Science and Technology, Beijing Normal University - Hong Kong Baptist University United International College (UIC), Zhuhai, Guangdong, China.
- PMID: 37338306
- DOI: 10.1002/bies.202200242
DNA extraction, playing an irreplaceable role in molecular biology as it is an essential step prior to various downstream biological analyses. Thus, the accuracy and reliability of downstream research outcomes depend largely on upstream DNA extraction methodology. However, with the advancement of downstream DNA detection techniques, the development of corresponding DNA extraction methods is lagging behind. The most innovative DNA extraction techniques are silica- or magnetic-based. Recent studies have demonstrated that plant fiber-based adsorbents (PF-BAs) have stronger DNA capturing ability than classic materials. Moreover, magnetic ionic liquid (MIL)-based DNA extraction has gathered attention lately, and extrachromosomal circular DNA (eccDNA), cell-free DNA (cfDNA), and microbial community DNA are current research hotspots. These require specific extraction methods, along with constant improvements in the way they are used. This review discusses the significance as well as the direction of innovation of DNA extraction methods to try to provide valuable references including current status and trends for DNA extraction.
Keywords: cell-free DNA; extrachromosomal circular DNA; magnetic ionic liquid; microbial community; plant fiber-based adsorbent.
© 2023 Wiley Periodicals LLC.
Publication types
- Ionic Liquids*
- Reproducibility of Results
- Ionic Liquids
- Open access
- Published: 12 February 2019
Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method
- Nadia Aboul-Ftooh Aboul-Maaty 1 &
- Hanaa Abdel-Sadek Oraby ORCID: orcid.org/0000-0001-9779-2953 1
Bulletin of the National Research Centre volume 43 , Article number: 25 ( 2019 ) Cite this article
86k Accesses
121 Citations
3 Altmetric
Metrics details
Reliable measurement of DNA concentration and purity is important for almost all molecular genetics studies. Different plant species have varying levels of polysaccharides, polyphenols, and other secondary metabolites which combine with nucleic acids during DNA isolation and further affect the quality of the extracted DNA. The current extraction protocol is based upon the conventional cetyl trimethylammonium bromide (CTAB) method with further modifications for the extraction of DNA from variable plant seeds and crops belonging to seven different orders. The principle modifications currently employed for DNA extraction involved the use of higher CTAB concentration and higher levels of 2-β-mercaptoethanol. Additionally, higher concentrations of sodium chloride and potassium acetate were added simultaneously with absolute ice cold isopropanol for the precipitation of DNA free from polysaccharides.
Results and conclusion
The prescribed modifications in the present method establish a quick and efficient standardized protocol for DNA extraction from different plant orders. The current extraction protocol, therefore, can be of great value for molecular analysis involving large numbers of different plant samples from different orders. These modifications consistently produced pure and high-quality DNA suitable for further molecular analysis. Successful PCR amplification with random amplified polymorphic DNA primer, NPTII gene, and the complete digestion of the isolated DNA with the HindIII restriction enzyme validated the quality of the isolated DNA. Moreover, it reflects the efficiency of the protocol and proves its suitability for further applications for the assessment of food safety, detection of genetically modified (GM) crops, and conservation of biodiversity.
Isolation and purification of DNA are a crucial step in DNA molecular techniques used in plant studies for the identification of genotypes, economical traits associated with genes of interest, and genetic diversity. Reliable measurement of DNA concentration and purity is also important for the assessment of food safety, especially with the increase of the global cultivation area of genetically modified (GM) crops (Ateş Sönmezoğlu and Keskin 2015 ). To facilitate protection of biodiversity and to guarantee rational use of these GM crops, sufficient measurements for purity, quality, and amount of DNA present in these products must be determined to comply with labeling regulation requirements.
DNA molecular techniques are mainly based on polymerase chain reaction (PCR) assay that requires isolation of genomic DNA of suitable purity. Various extraction protocols have been established in order to isolate pure and intact whole genomic DNA from plant tissues (Saghai-Maroof et al. 1984 ; Doyle and Doyle 1990 ; Scott and Playford 1996 ; Sharma et al. 2000 ; Pirttilä et al. 2001 ; Shepherd et al. 2002 ; Mogg and Bond 2003 ; Haymes 1996 ).
However, many difficulties have been reported for isolating good-quality DNA from plants (Novaes et al. 2009 ; Silva 2010 ; Moreira and Oliveira 2011 ). These difficulties were attributed to the fact that different plant species have varying levels of polysaccharides, polyphenols, and other secondary metabolites. These components are usually hindering the process of DNA purification and its further use in molecular studies (Khanuja et al. 1999 ). These plant components have a similar structure of nucleic acids that allow secondary metabolites and polysaccharides to interfere with total DNA isolation (Shioda and Marakami-Muofushi 1987 ). They strongly combine with nucleic acids during DNA isolation and affect the quality of the extracted DNA from higher plants (Scott and Playford 1996 ). These metabolites also affect the quantity and purity of the isolated nucleic acids (Porebski et al. 1997 ). The removal of such contaminants needs complicated and time-consuming protocols. A single DNA isolation protocol is not likely to be applicable for all the plant tissues (Loomis 1974 ). Most of the cetyl trimethylammonium bromide (CTAB)-based protocols used for the extraction of DNA were tailored according to the internal components of each single plant species (Wang et al. 2012 ; Moreira and Oliveira 2011 ).
The present work describes an inexpensive CTAB-based method with modifications for the extraction of high-quality genomic DNA from 19 different plant seeds and crops belong to seven different plant orders. These plant samples are rich in proteins, polysaccharides, and polyphenols. In comparison, we used the classical protocol of Doyle and Doyle ( 1990 ) for isolation of DNA from the same samples. In order to validate the quality of the DNA extracted by the modified protocol, PCR amplification of genomic DNA extracted from different plant seeds applying the two utilized protocols was carried out using random amplified polymorphic DNA (RAPD). PCR amplification of neomycin phosphotransferase gene (nptII) was used to evaluate the efficacy of the present protocol to produce good-quality DNA suitable for detection of genetically modified crops.
Materials and methods
Plant materials.
Twenty-seven plant samples were purchased locally from plant seed suppliers in Egypt (Table 1 ). They were chosen to be enrolled in this study because they have varying amounts of polysaccharides, proteins, and polyphenols and they belong to seven different orders. These plant samples were mainly imported from different countries distributed in Europe, America, and Asia. Additionally, four animal diet samples (D1, D2, D3, and D4) were also purchased from different suppliers. These four diet samples contain mixtures of soybean and corn.
3× extraction buffer containing: 3% CTAB ( w / v ), 1.4 M NaCl, 0.8 M Tris-HCl pH 8.0, 0.5 M EDTA pH 8.0 (autoclaved)
0.3% 2-β-Mercaptoethanol.
Chloroform:isoamyl alcohol (24:1 v / v ).
3 M potassium acetate
Ice cold 100% isopropyl alcohol
70% ethanol
1× TE buffer (10 mM Tris-HCl, pH 8.0; 1 mM EDTA, pH 8.0, autoclaved).
Agarose (molecular grade)
Modified DNA extraction protocol
Preheat the 3× extraction buffer in water bath at 65 °C. Add 0.3% 2-β-mercaptoethanol to the 3× CTAB extraction buffer immediately before use.
Grind 50 mg of plant samples into powder in liquid nitrogen using pre chilled mortar and pestle. While still in the mortar, add 800 μl of the preheated 3× CTAB extraction buffer to the grinded plant samples and swirl gently to mix using the pestle.
Transfer the sample mixture to a 2-ml microcentrifuge tube, incubate in water bath at 60–65 °C for 1 h, mix gently every 20 min by inverting the tube for 20 times each, then cool down to the room temperature.
Add an equal volume of chloroform:isoamyl alcohol (24:1 v / v ) and mix by slight inversion.
Centrifuge at 13,000 rpm for 15 min at room temperature (RT).
Using a wide bore pipet, carefully transfer the upper aqueous phase, which contains the DNA, to a new 1.5-ml eppendorf tube.
Repeat the extraction steps (iv–vi), when necessary until the upper aqueous phase is clear.
Estimate the volume of the aqueous phase (approximately 700 μl) then add half this volume (350 μl) of 6 M NaCl and mix well. Successively, add 1/10 the volume (70 μl) 3 M potassium acetate and simultaneously mix with 500 μl ice cold 100% isopropyl alcohol (approximately two thirds the volume of the aqueous phase). Invert gently to precipitate DNA until the formation of DNA threads.
Incubate at − 20 °C for 30 min.
Centrifuge at 13,000 rpm for 5 min, discard supernatant.
Invert the tube containing the DNA pellet on tissue paper to complete draining off the supernatant.
Wash DNA pellet with 500 μl of 70% ethanol and invert once (to dissolve residual salts and to increase purity of the DNA).
Centrifuge at 13,000 rpm for 5 min.
Discard 70% alcohol from tubes. invert the on filter paper, and allow tubes containing pellet to air dry at room temperature for 15 min.
Re-suspend the DNA pellet in 50 μl 1× TE buffer. Incubate the DNA at 50 °C for 1 to 2 h to ensure complete re-suspension.
Store at − 20 °C till further use.
Quantitative and qualitative analysis of DNA extracted by established CTAB method and modified protocol
Dna concentration, purity, and quality.
DNA concentration was determined spectrophotometrically at 260 nm (A 260 ) absorption using NanoDrop1000 (Thermo Scientific). Purity of DNA from protein and polysaccharide contamination (Wilson and Walker 2005 ) was assessed by estimating the absorbance ratio at A 260 /A 280 and A 260 /A 230 respectively. The quality of the extracted DNA using both protocols was also evaluated by electrophoresis separation for all DNA samples on 0.8% agarose gel stained with ethidium bromide (1 μg/ml).
DNA digestion analysis
HindIII restriction enzyme was used to digest the DNA samples according to the procedure of Fang and colleagues ( 1992 ). Approximately 20 μg of genomic DNA was digested separately for 1 h at 37 °C with HindIII restriction enzyme (Amersham Pharmacia Biotech. UK Ltd). All stained electrophoresis separation matrices for PCR amplification and both extracted and digested DNA samples were resolved by SYNGENE Bio Imaging Gel Documentation System (UK).
Random amplified polymorphic DNA analysis
PCR amplification of genomic DNA extracted from different plant seeds applying the two protocols utilized was carried out using random amplified polymorphic DNA (RAPD) decamer primer (OPZ-09) that was synthesized by Operon Primer Kits (Operon, USA). The primer sequence is 5′-CAGCACTGAC-3′. PCR was performed for all samples according to the method described by Devi and colleagues ( 2013 ).
Detection of genetically modified (GM) crops
The efficacy of the present protocol to produce good-quality DNA suitable for detection of genetically modified crops was also assessed. The isolated genomic DNA from different plant samples by means of the present protocol and the conventional method was used as a template for PCR amplification of neomycin phosphotransferase gene (nptII), which is utilized as a selectable marker gene in the transformation processes. The existence of NPTII (173 bp target) was investigated in the plant seeds enrolled in the present work, using specific primers for this gene (F: 5′-GGATCTCCTGTCATCT-3′ and R: 5′-GGATCTCCTGTCATCT-3′). The PCR amplification was carried out in a 25-μl reaction mixture containing 12.75 μl of DNase free water, 100 ng template DNA (2 μl), 200 μM of each dNTP (2.5 μl), 2.5 pmol of each primer (2.5 μl), and 2.5 units of taq DNA polymerase (0.25 μl) in a reaction buffer (2.5 μl) containing 75 mM Tris-HCl, pH 8.0, 2 mM MgCl 2 , 50 mM KCl, 20 mM (NH 4 ) 2 SO 4 , and 0.001% BSA.
PCR amplifications were performed in a TM Thermal cycler (MJ Research PTC-100 thermocycler) programmed to perform an initial denaturation step of 98 °C for 2 min, followed by 40 cycles consisting of 30 s at 95 °C for denaturation, 45 s at annealing temperature (50 °C), and 30 s at 72 °C for extension. A final extension step of 7 min at 72 °C was performed. Following completion of the cycling reaction, 2 μl of a loading dye (bromophenol blue) was added to 10 μl of each reaction product and separated by 2% agarose gel electrophoresis stained with 1 μg/ml ethidium bromide. PCR products were analyzed, using SYNGENE Bio Imaging Gel Documentation System, for the presence of a fluorescent band of the expected base pair (bp) size (173 bp).
Applying the present standardized method, the extracted DNA concentrations varied with the different plant species used in the present work (Table 2 ). The yield of isolated DNA ranged from 2.238 ηg/mg of seeds in case of Cucurbitales maxima to 24.957 ηg/mg of seeds in the case of Lupinus lupinus. The other classical CTAB method employed (Doyle and Doyle 1990 ) also produced comparable range of DNA concentration (Table 2 ), yet with less purity in most cases. Most of DNA samples extracted by the original CTAB method had A 260 /A 280 ratio below 1.8, while the A 260 /A 280 ratios ranged from 2.08 to 2.23 in DNA samples extracted by our modified protocol.
The quality of the total DNA extracted by the present protocol, from different plant species, was also evaluated by electrophoresis separation. Results showed intense bands very close to the gel wells (Fig. 1 , upper lane). Genomic DNA extracted by the CTAB method of Doyle and Doyle ( 1990 ) from the same samples did not produce distinct or intact bands (Fig. 1 , lower lane). The NanoDrop spectrophotometer measurement profile showed a single absorbance peak at 260 nm in DNA samples extracted by our standardized protocol. Figure 2 shows an example of a NanoDrop measurement profile of extracted genomic DNA from Glycine max sample using our protocol. DNA samples extracted by the present modified extraction protocol were efficiently digested with the HindIII restriction enzyme (Fig. 3 ).

Quality of extracted DNA. Quality of DNA extracted from some of the plant samples using both DNA extraction methods. Electrophoresis separation was performed on 0.8% agarose gel matrix, stained with ethidium bromide. The upper lane is for samples extracted by the standardized method after modification; the lower lane is for the same samples extracted by the conventional method. M is a molecular marker (100 bp)

NanoDrop measurement profile. NanoDrop measurement profile of the extracted genomic DNA from the Glycine max sample using the modified protocol

DNA digested with HindIII enzyme. DNA samples extracted by the standardized method and digested with HindIII enzyme. Digested products were separated in 1.5% agarose gel stained with ethidium bromide, in 1 × TAE buffer. The digested products were visualized by UV fluorescence. M is a molecular marker (50 bp)
PCR amplification with RAPD primer (OPZ-09) showed clear and well-differentiated band patterns (Fig. 4 ) in case of DNA samples extracted by the present DNA extraction protocol, whereas genomic DNA extracted by the other method from the same plant seed samples was rather difficult to be amplified (Fig. 4 ). Figure 5 shows the differences in the quality of the PCR amplification products of nptII (173 bp target) in plant samples which were extracted by both the conventional method and by the modified protocol.

PCR amplification with RAPD primer (OPZ-09). PCR amplification of OPZ-09 primer and electrophoresis separation by 1.5% agarose gel of some DNA samples extracted by the standardized method (upper lane) and the classical method (lower lane). M is for molecular marker (50 bp)

PCR amplification with nptII for detection of GM crops. Quality of the amplification of nptII (173 bp target) in the representative of DNA samples which were extracted by the conventional method (upper lane) and the standardized protocol (lower lane)
The extraction of DNA from plant seeds is an essential step for satisfactory results in molecular studies particularly those involving plant genetics (Junior et al. 2016 ). Different seeds belonging to related genera or different orders contain many components with variable complexities that badly interfere with purity of the extracted DNA and molecular investigations following isolation procedures (Porebski et al. 1997 ; Ribeiro and Lovato 2007 ).
To insure isolation of DNA with better yield and quality from seeds of diverse plant orders, we implemented several steps in the present modified protocol. Liquid nitrogen was used to break the cell wall and disrupt the cell membrane (Clark 1997 ) while keeping cellular enzymes and other undesired chemicals deactivated, thus reducing shearing and damaging of the DNA. Other methods used for disrupting plant tissues, such as digestion with pectinase and cellulose (Manen et al. 2005 ), are not as reproducible or accurate as the use of liquid nitrogen.
High concentration of the 3× CTAB was also used to disrupt the cells and nuclear membranes in order to expose the genetic components (Amani et al. 2011 ). In the present modified method, the 3× CTAB buffer also contains the highest recommended concentration level (0.3%) of 2-β-mercaptoethanol which successfully removed polyphenols (Horne et al. 2004 ; Li et al. 2007 ) giving rise a clear translucent DNA pellet. The CTAB extraction buffer also includes 1.4 M of NaCl which improved the quality of the extracted DNA (Sahu et al. 2012 ).
To remove the remaining polysaccharides during DNA extraction from all plant samples included in the present work, a modification for the precipitation of DNA was also performed by increasing the concentration of sodium chloride and potassium acetate. The concentration of NaCl varied with plant species in a range between 0.7 M (Clark 1997 ) and 6 M (Aljanabi et al. 1999 ; Moreira and Oliveira 2011 ). In the present standardized protocol, we used 6 M NaCl (Moreira and Oliveira 2011 ) and 3 M potassium acetate (Paterson et al. 1993 ). These modifications successfully removed polysaccharides impurities from DNA extracted by this modified protocol from all plant samples and produced pure and high-quality DNA suitable for further molecular analysis. Proteins, most lipids, and cellular debris were removed by binding with non-aqueous compounds and precipitated during the chloroform-isoamyl alcohol step.
Longer incubation of the extracted DNA at − 20 °C also enhanced precipitation of DNA. In general, the quantity and quality of isolated DNA depend on precipitation temperature and duration (Michiels et al. 2003 ). Low-temperature precipitation employed in the present modified protocol increased DNA yield. Extracted DNA were re-suspended in minimum amount of 1× TE buffer since the presence of chelating agents in TE buffer can affect the PCR and other molecular analysis of the extracted DNA.
The method employed in the present work proved to be successful and applicable for extraction of DNA with high yield and purity from 19 different plant species that belong to seven different plant orders. The matrix variation effects on the purity and quality of the isolated genomic DNA were minimized by using the same plant samples as starting materials for both protocols employed in the present investigation.
Electrophoresis separation of DNA extracted by the present protocol showed intense bands very close to the gel wells (Fig. 1 , upper lane) signifying high degree of purity and intact DNA. It is known that the presence of smear could be a sign of degradation of the extracted DNA which easily affects the quality of the subsequent molecular application results (Devi et al. 2013 ).
DNA samples extracted by the present protocol were assessed for successful PCR amplification with RAPD primer (OPZ-09). The presence of clear and well-differentiated band patterns (Fig. 4 ) reflects the efficiency of the protocol to produce genomic DNA with high purity suitable for molecular studies that based on PCR techniques (Devi et al. 2013 ).
Purification of DNA is also an important step for analyzing and measuring genetically modified (GM) food products (Ateş Sönmezoğlu and Keskin 2015 ). The DNA extracted by our standardized protocol yielded detectable and reproducible bands for NPTII (173 bp target) proving its suitability for PCR amplification as well as for the identification of GM crops using the PCR assay.
The A 260 /A 280 purity ratio is an important measure for estimating the polyphenol contamination levels of the extracted DNA. Ratios of A 260 /A 280 below 1.8 render the extracted DNA inappropriate for molecular investigations (Sambrook and Russell 2001 ). Therefore, higher level of 2-β-mercaptoethanol (0.3%) used in the present standardized method successfully removed polyphenols giving rise to translucent final DNA pellets (Suman et al. 1999 ).
In the present modified CTAB-based protocol, although the RNase A enzyme was not used during isolation and purification of DNA, the ratios of absorption A 260 /A 280 of the extracted DNA (Table 2 ) were higher than the recommended optimal limit of DNA purity (Sambrook and Russell 2001 ). Similar results were also observed by Sambrook and Russell ( 2001 ) which were taken to be associated with RNA contamination. In our case, the resulted intact DNA bands, very close to the wells (Fig. 1 , upper lane), indicated high purity of the extracted DNA with no RNA contamination, particularly that the recommended and the most accurate way to determine RNA contamination is to run the sample on an agarose gel where another band of the RNA, if present, will be visible in the gel (Wang et al. 2012 ). Therefore, the higher ratios of absorption A 260 /A 280 in our case may be attributed to slight changes in the pH of the extracted samples (Wilfinger et al. 1997 ).
Polysaccharide contamination was also assessed (Table 2 ) by estimating the absorbance ratio A 260 /A 230 as a secondary measure of nucleic acid purity (Wilson and Walker 2005 ). This ratio is important to evaluate the level of salt residues in the purified DNA. It is recommended to be greater than 1.5 and preferably close to 1.8. The reported values of A 260 /A 230 ratio in most of the DNA plant samples extracted by the present modified protocol are higher than those of the DNA samples extracted by the other classical method.
The principle modifications currently employed for DNA extraction involved the use of higher CTAB concentration and higher levels of 2-β-mercaptoethanol. Additionally, higher concentrations of sodium chloride and potassium acetate were added simultaneously with absolute ice cold isopropanol for the precipitation of DNA free from polysaccharides.
The prescribed modifications in the present method establish a quick and efficient standardized protocol for DNA extraction from different plant orders. These modifications consistently produced pure and high-quality DNA suitable for further molecular analysis. The DNA standardized extraction protocol presented here is important for the assessment of food safety, detection of genetically modified crops, and biodiversity conservation. Therefore, it is of great value for molecular analysis involving large number of different plant samples.
Abbreviations
- Cetyl trimethylammonium bromide
Deoxyribonucleic acid
Genetically modified organisms
Hydrochloric acid
Sodium chloride
Neomycinphosphotransferase II gene
Optical density
Random amplified polymorphic DNA
Aljanabi MS, Forget L, Dookun A (1999) An improved and rapid protocol for the isolation of polysaccharide and polyphenol free sugarcane DNA. Plant Mol Biol Rep 17:1–8
Article Google Scholar
Amani J, Kazemi R, Abbasi AR, Salmanian AH (2011) A simple and rapid leaf genomic DNA extraction method for polymerase chain reaction analysis. Iran J Biotech 9:69
CAS Google Scholar
Ateş Sönmezoğlu Ö, Keskin H (2015) Determination of genetically modified corn and soy in processed food products. J App Biol Biotech 3:032
Google Scholar
Clark MS (ed) (1997) Plant molecular biology- a laboratory manual. Springer, New York, pp 305–328
Devi KD, Punyarani K, Singh S, Devi HS (2013) An efficient protocol for total DNA extraction from the members of order Zingiberales - suitable for diverse PCR based downstream applications. Springer Plus 2:669. https://doi.org/10.1186/2193-180-2-669
Article PubMed Google Scholar
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13
Fang G, Hammar S, Grumet R (1992) A quick and inexpensive method for removing polysaccharides from plant genomic DNA. BioTechniques 13:52–56
CAS PubMed Google Scholar
Haymes KM (1996) Mini-prep method suitable for a plant breeding program. Plant Mol Biol Rep 14:280
Article CAS Google Scholar
Horne EC, Kumpatla SP, Patterson MG, Thompson SA (2004) Improved high-throughput sunflower and cotton genomic DNA extraction and PCR fidelity. Plant Mol Biol Rep. 22:83
Júnior CDS, Teles NMM, Luiz DP, Isabel TF (2016) DNA Extraction from Seeds. In: Micic M, editor. Sample Preparation Techniques for Soil, Plant, and Animal Samples. Springer Protocols Handbooks. Humana Press, New York, pp.265-276. https://doi.org/10.1007/978-1-4939-3185-9_18
Khanuja SPS, Shasany AK, Darokar MP, Kumar S (1999) Rapid isolation of DNA from dry and fresh samples of plants producing large amounts of secondary metabolites and essential oils. Plant Mol Biol Rep. 17:1
Li JT, Yang J, Chen DC, Zhang XL, Tang ZS (2007) An optimized mini-preparation method to obtain high-quality genomic DNA from mature leaves of sunflower. Genet Mol Res 6:1064
Loomis MD (1974) Overcoming problems of phenolics and quinones in the isolation of plant enzymes and organelles. Methods Enzymol 31:528
Manen JF, Sinitsyna O, Aeschbach L, Markov AV, Sinitsyn A (2005) A fully automatable enzymatic method for DNA extraction from plant tissues. BMC Plant Biol 5:23
Michiels A, Van den Ende W, Tucker M, Van Riet L (2003) Extraction of high-quality genomic DNA from latex-containing plants. Anal Biochem 315:85
Mogg RJ, Bond JM (2003) A cheap, reliable and rapid method of extracting high-quality DNA from plants. Mol Ecol Notes 3:666
Moreira PA, Oliveira DA (2011) Leaf age affects the quality of DNA extracted from Dimorphandra mollis (Fabaceae), atropical tree species from the Cerrado region of Brazil. Genet Mol Res 10:353
Novaes RML, Rodrigues JG, Lovato MB (2009) An efficient protocol for tissue sampling and DNA isolation from the stem bark of Leguminosae trees. Genet Mol Res 8:86–96
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton ( Gossypium spp. ) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep. 11:122
Pirttilä MA, Hirsikorpi M, Kämäräinen T, Jaakola L, Hohtola A (2001) DNA isolation methods for medicinal and aromatic plants. Plant Mol Biol Rep. 19:273
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep. 15:8–15
Ribeiro RA, Lovato MB (2007) Comparative analysis of different DNA extraction protocols in fresh and herbarium specimens of the genus Dalbergia. Genet Mol Res 6:173
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA sepacer-length polymorphism in barley: Mendelian inheritance, chromosalmal localtionk, and population dynamic. Proc Natl Acad Sci U S A 81:8014
Article ADS CAS Google Scholar
Sahu SK, Thangaraj M, Kathiresan KDNA (2012) Extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. Mol Biol 12:1
Sambrook J, Russell DW (2001) Molecular Cloning. A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York
Scott KD, Playford J (1996) DNA extraction technique for PCR in rain forest plant species. Bio Techniques 20:974
Sharma KK, Lavanya M, V A (2000) A method for isolation and purification of peanut genomic DNA suitable for analytical applications. Plant BioTechniques Rep 18:393a
Shepherd M, Cross M, Stokoe RL, Scott LJ (2002) High-throughput DNA extraction from forest trees. Plant Mol Biol Rep. 20:425
Shioda M, Marakami-Muofushi K (1987) Selective inhibition of DNA polymerase by a polysaccharide purified from slime of Physarum polycephalum . Biochem Biophys Res Commun 146:61–66
Silva MN (2010) Extraction of genomic DNA from leaf tissues of mature native species of the Cerrado. Rev. Árvore 34:973–978
Suman PSK, Ajit KS, Darokar MP, Sushil K (1999) Rapid isolation of DNA from dry and fresh samples of plants producing large amounts of secondary metabolites and essential oils. Plant Mol Biol Rep. 17:1
Wang X, Xiao H, Zhao X, Li C, Ren J, Wang F , Pang L. Isolation of high-quality DNA from a desert plant Reaumuria soongorica, genetic diversity in plants, Mahmut Caliskan (Ed.), ISBN: 978–953–51-0185-7, InTech; 2012. https://doi.org/10.5772/38367
Wilfinger WW, Mackey K, Chomczynski P (1997) Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. BioTechniques 22:474–481
Wilson K, Walker J (2005) Principles and techniques of biochemistry and molecular biology. University Press, Cambridge
Book Google Scholar
Download references
Acknowledgements
Not applicable
The authors declare that this work was funded by the National Research Centre in Egypt (the 11th Research Project Plan, 2016-2019, Project ID: 11040201).
Availability of data and materials
We declare that all data generated or analyzed during this study are included in this article.
Author information
Authors and affiliations.
Cell Biology Department, Genetic Engineering and Biotechnology Research Division, National Research Centre, Cairo, Dokki, 11622, Egypt
Nadia Aboul-Ftooh Aboul-Maaty & Hanaa Abdel-Sadek Oraby
You can also search for this author in PubMed Google Scholar
Contributions
NA-M made substantial contributions to conception and design of the work, involved in conducting the practical section of the work, and also involved in drafting the manuscript. HO made substantial contributions to conception, planning of the work, analysis, and interpretation of results and also involved in drafting the manuscript and revising it critically for important intellectual content, as well as gave the final approval of the version to be published. Each author has participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Both authors read and approved the final manuscript.
Corresponding author
Correspondence to Hanaa Abdel-Sadek Oraby .
Ethics declarations
Ethics approval and consent to participate, consent for publication, competing interests.
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/ ), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Reprints and permissions
About this article
Cite this article.
Aboul-Maaty, N.AF., Oraby, H.AS. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull Natl Res Cent 43 , 25 (2019). https://doi.org/10.1186/s42269-019-0066-1
Download citation
Received : 15 August 2018
Accepted : 31 January 2019
Published : 12 February 2019
DOI : https://doi.org/10.1186/s42269-019-0066-1
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Isolation of nucleic acid
- Plant seeds
- Molecular techniques
- Quality of nucleic acids
- Plants’ genomic DNA
- GMO detection
Cellular activity hints that recycling is in our DNA
New research shows that 'spliceosomes' might reinsert problematic gene sequences after removing them.
Although you may not appreciate them, or have even heard of them, throughout your body, countless microscopic machines called spliceosomes are hard at work. As you sit and read, they are faithfully and rapidly putting back together the broken information in your genes by removing sequences called "introns" so that your messenger RNAs can make the correct proteins needed by your cells.
Introns are perhaps one of our genome's biggest mysteries. They are DNA sequences that interrupt the sensible protein-coding information in your genes, and need to be "spliced out." The human genome has hundreds of thousands of introns, about 7 or 8 per gene, and each is removed by a specialized RNA protein complex called the "spliceosome" that cuts out all the introns and splices together the remaining coding sequences, called exons. How this system of broken genes and the spliceosome evolved in our genomes is not known.
Over his long career, Manny Ares, UC Santa Cruz distinguished professor of molecular, cellular, and developmental biology, has made it his mission to learn as much about RNA splicing as he can.
"I'm all about the spliceosome," Ares said. "I just want to know everything the spliceosome does -- even if I don't know why it is doing it."
In a new paper published in the journal Genes and Development , Ares reports on a surprising discovery about the spliceosome that could tell us more about the evolution of different species and the way cells have adapted to the strange problem of introns. The authors show that after the spliceosome is finished splicing the mRNA, it remains active and can engage in further reactions with the removed introns.
This discovery provides the strongest indication we have so far that spliceosomes could be able to reinsert an intron back into the genome in another location. This is an ability that spliceosomes were not previously believed to possess, but which is a common characteristic of "Group II introns," distant cousins of the spliceosome that exist primarily in bacteria.
The spliceosome and Group II introns are believed to share a common ancestor that was responsible for spreading introns throughout the genome, but while Group II introns can splice themselves out of RNA and then directly back into DNA, the "spliceosomal introns" that are found in most higher-level organisms require the spliceosome for splicing and were not believed to be reinserted back into DNA. However, Ares's lab's finding indicates that the spliceosome might still be reinserting introns into the genome today. This is an intriguing possibility to consider because introns that are reintroduced into DNA add complexity to the genome, and understanding more about where these introns come from could help us to better understand how organisms continue to evolve.
Building on an interesting discovery
An organism's genes are made of DNA, in which four bases, adenine (A), cytosine (C), guanine (G) and thymine (T) are ordered in sequences that code for biological instructions, like how to make specific proteins the body needs. Before these instructions can be read, the DNA gets copied into RNA by a process known as transcription, and then the introns in that RNA have to be removed before a ribosome can translate it into actual proteins.
The spliceosome removes introns using a two-step process that results in the intron RNA having one of its ends joined to its middle, forming a circle with a tail that looks like a cowboy's "lariat," or lasso. This appearance has led to them being named "lariat introns." Recently, researchers at Brown University who were studying the locations of the joining sites in these lariats made an odd observation -- some introns were actually circular instead of lariat shaped.
This observation immediately got Ares's attention. Something seemed to be interacting with the lariat introns after they were removed from the RNA sequence to change their shape, and the spliceosome was his main suspect.
"I thought that was interesting because of this old, old idea about where introns came from," Ares said. "There is a lot of evidence that the RNA parts of the spliceosome, the snRNAs, are closely related to Group II introns."
Because the chemical mechanism for splicing is very similar between the spliceosomes and their distant cousins, the Group II introns, many researchers have theorized that when the process of self-splicing became too inefficient for Group II introns to reliably complete on their own, parts of these introns evolved to become the spliceosome. While Group II introns were able to insert themselves directly back into DNA, however, spliceosomal introns that required the help of spliceosomes were not thought to be inserted back into DNA.
"One of the questions that was sort of missing from this story in my mind was, is it possible that the modern spliceosome is still able to take a lariat intron and insert it somewhere in the genome?" Ares said. "Is it still capable of doing what the ancestor complex did?"
To begin to answer this question, Ares decided to investigate whether it was indeed the spliceosome that was making changes to the lariat introns to remove their tails. His lab slowed the splicing process in yeast cells, and discovered that after the spliceosome released the mRNA that it had finished splicing introns from, it hung onto intron lariats and reshaped them into true circles. The Ares lab was able to reanalyze published RNA sequencing data from human cells and found that human spliceosomes also had this ability.
"We are excited about this because while we don't know what this circular RNA might do, the fact that the spliceosome is still active suggests it may be able to catalyze the insertion of the lariat intron back into the genome," Ares said.
If the spliceosome is able to reinsert the intron into DNA, this would also add significant weight to the theory that spliceosomes and Group II introns shared a common ancestor long ago.
Testing a theory
Now that Ares and his lab have shown that the spliceosome has the catalytic ability to hypothetically place introns back into DNA like their ancestors did, the next step is for the researchers to create an artificial situation in which they "feed" a DNA strand to a spliceosome that is still attached to a lariat intron and see if they can actually get it to insert the intron somewhere, which would present "proof of concept" for this theory.
If the spliceosome is able to reinsert introns into the genome, it is likely to be a very infrequent event in humans, because the human spliceosomes are in incredibly high demand and therefore do not have much time to spend with removed introns. In other organisms where the spliceosome isn't as busy, however, the reinsertion of introns may be more frequent. Ares is working closely with UCSC Biomolecular Engineering Professor Russ Corbett-Detig, who has recently led a systematic and exhaustive hunt for new introns in the available genomes of all intron-containing species that was published in the journal Proceedings of the National Academy of Sciences (PNAS) last year.
The paper in PNAS showed that intron "burst" events far back in evolutionary history likely introduced thousands of introns into a genome all at once. Ares and Corbett-Detig are now working to recreate a burst event artificially, which would give them insight into how genomes reacted when this happened.
Ares said that his cross-disciplinary partnership with Corbett-Detig has opened the doors for them to really dig into some of the biggest mysteries about introns that would probably be impossible for them to understand fully without their combined expertise.
"It is the best way to do things," Ares said. "When you find someone who has the same kind of questions in mind but a different set of methods, perspectives, biases, and weird ideas, that gets more exciting. That makes you feel like you can break out and solve a problem like this, which is very complex."
- Human Biology
- Back and Neck Pain
- Biochemistry Research
- New Species
- Evolutionary Biology
- Genetic code
- Human genome
- DNA microarray
Story Source:
Materials provided by University of California - Santa Cruz . Original written by Rose Miyatsu. Note: Content may be edited for style and length.
Journal Reference :
- Manuel Ares, Haller Igel, Sol Katzman, John P. Donohue. Intron lariat spliceosomes convert lariats to true circles: implications for intron transposition . Genes & Development , 2024; DOI: 10.1101/gad.351764.124
Cite This Page :
Explore More
- Nature's 3D Printer: Bristle Worms
- Giant ' Cotton Candy' Planet
- A Young Whale's Journey
- No Inner Voice Linked to Poorer Verbal Memory
- Bird Flu A(H5N1) Transmitted from Cow to Human
- Universe's Oldest Stars in Our Galactic Backyard
- Polygenic Embryo Screening for IVF: Opinions
- VR With Cinematoghraphics More Engaging
- 2023 Was the Hottest Summer in 2000 Years
- Fastest Rate of CO2 Rise Over Last 50,000 Years
Trending Topics
Strange & offbeat.
MIT Technology Review
- Newsletters
Google DeepMind’s new AlphaFold can model a much larger slice of biological life
AlphaFold 3 can predict how DNA, RNA, and other molecules interact, further cementing its leading role in drug discovery and research. Who will benefit?
- James O'Donnell archive page
Google DeepMind has released an improved version of its biology prediction tool, AlphaFold, that can predict the structures not only of proteins but of nearly all the elements of biological life.
It’s a development that could help accelerate drug discovery and other scientific research. The tool is currently being used to experiment with identifying everything from resilient crops to new vaccines.
While the previous model, released in 2020, amazed the research community with its ability to predict proteins structures, researchers have been clamoring for the tool to handle more than just proteins.
Now, DeepMind says, AlphaFold 3 can predict the structures of DNA, RNA, and molecules like ligands, which are essential to drug discovery. DeepMind says the tool provides a more nuanced and dynamic portrait of molecule interactions than anything previously available.
“Biology is a dynamic system,” DeepMind CEO Demis Hassabis told reporters on a call. “Properties of biology emerge through the interactions between different molecules in the cell, and you can think about AlphaFold 3 as our first big sort of step toward [modeling] that.”
AlphaFold 2 helped us better map the human heart , model antimicrobial resistance , and identify the eggs of extinct birds , but we don’t yet know what advances AlphaFold 3 will bring.
Mohammed AlQuraishi, an assistant professor of systems biology at Columbia University who is unaffiliated with DeepMind, thinks the new version of the model will be even better for drug discovery. “The AlphaFold 2 system only knew about amino acids, so it was of very limited utility for biopharma,” he says. “But now, the system can in principle predict where a drug binds a protein.”
Isomorphic Labs, a drug discovery spinoff of DeepMind, is already using the model for exactly that purpose, collaborating with pharmaceutical companies to try to develop new treatments for diseases, according to DeepMind.
AlQuraishi says the release marks a big leap forward. But there are caveats.
“It makes the system much more general, and in particular for drug discovery purposes (in early-stage research), it’s far more useful now than AlphaFold 2,” he says. But as with most models, the impact of AlphaFold will depend on how accurate its predictions are. For some uses, AlphaFold 3 has double the success rate of similar leading models like RoseTTAFold. But for others, like protein-RNA interactions, AlQuraishi says it’s still very inaccurate.
DeepMind says that depending on the interaction being modeled, accuracy can range from 40% to over 80%, and the model will let researchers know how confident it is in its prediction. With less accurate predictions, researchers have to use AlphaFold merely as a starting point before pursuing other methods. Regardless of these ranges in accuracy, if researchers are trying to take the first steps toward answering a question like which enzymes have the potential to break down the plastic in water bottles, it’s vastly more efficient to use a tool like AlphaFold than experimental techniques such as x-ray crystallography.
A revamped model
AlphaFold 3’s larger library of molecules and higher level of complexity required improvements to the underlying model architecture. So DeepMind turned to diffusion techniques, which AI researchers have been steadily improving in recent years and now power image and video generators like OpenAI’s DALL-E 2 and Sora. It works by training a model to start with a noisy image and then reduce that noise bit by bit until an accurate prediction emerges. That method allows AlphaFold 3 to handle a much larger set of inputs.
That marked “a big evolution from the previous model,” says John Jumper, director at Google DeepMind. “It really simplified the whole process of getting all these different atoms to work together.”
It also presented new risks. As the AlphaFold 3 paper details, the use of diffusion techniques made it possible for the model to hallucinate, or generate structures that look plausible but in reality could not exist. Researchers reduced that risk by adding more training data to the areas most prone to hallucination, though that doesn’t eliminate the problem completely.
Restricted access
Part of AlphaFold 3’s impact will depend on how DeepMind divvies up access to the model. For AlphaFold 2, the company released the open-source code , allowing researchers to look under the hood to gain a better understanding of how it worked. It was also available for all purposes, including commercial use by drugmakers. For AlphaFold 3, Hassabis said, there are no current plans to release the full code. The company is instead releasing a public interface for the model called the AlphaFold Server , which imposes limitations on which molecules can be experimented with and can only be used for noncommercial purposes. DeepMind says the interface will lower the technical barrier and broaden the use of the tool to biologists who are less knowledgeable about this technology.
Artificial intelligence
Sam altman says helpful agents are poised to become ai’s killer function.
Open AI’s CEO says we won’t need new hardware or lots more training data to get there.
What’s next for generative video
OpenAI's Sora has raised the bar for AI moviemaking. Here are four things to bear in mind as we wrap our heads around what's coming.
- Will Douglas Heaven archive page
Is robotics about to have its own ChatGPT moment?
Researchers are using generative AI and other techniques to teach robots new skills—including tasks they could perform in homes.
- Melissa Heikkilä archive page
An AI startup made a hyperrealistic deepfake of me that’s so good it’s scary
Synthesia's new technology is impressive but raises big questions about a world where we increasingly can’t tell what’s real.
Stay connected
Get the latest updates from mit technology review.
Discover special offers, top stories, upcoming events, and more.
Thank you for submitting your email!
It looks like something went wrong.
We’re having trouble saving your preferences. Try refreshing this page and updating them one more time. If you continue to get this message, reach out to us at [email protected] with a list of newsletters you’d like to receive.
share this!
May 9, 2024
This article has been reviewed according to Science X's editorial process and policies . Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
New DNA origami technique promises advances in medicine
by University of Portsmouth
. DOI: 10.1021/jacs.4c03413")
A new technique in building DNA structures at a microscopic level has the potential to advance drug delivery and disease diagnosis, a study suggests.
A team of scientists, from the universities of Portsmouth and Leicester in the UK, has developed an innovative way to customize and strengthen DNA origami.
DNA origami is the method of creating nanostructures with remarkable precision using DNA strands as building blocks. However, these structures are delicate and can fall apart easily under biological conditions, like changes in temperature or exposure to certain enzymes found in living organisms.
In a paper, published in the Journal of the American Chemical Society , researchers have presented a unique way to make the origami structures stronger and more versatile in a one-pot reaction, via a process known as triplex-directed photo-cross-linking.
By strategically modifying DNA strands during the design process , they were able to introduce additional nucleotide sequences—which are the basic building blocks of DNA—that serve as attachment points for functional molecules.
Attachment of the molecules was achieved by using triplex-forming oligonucleotides carrying a cross-linking agent. They then used a chemical process involving UVA light to permanently link these molecules to the DNA shapes.
A particular benefit of this approach is the generation of "super-staples" that act to weave the structure together. The paper says cross-linking to regions outside of the origami core dramatically reduces the structure's sensitivity to heat and disassembly by enzymes.
Senior author, Dr. David Rusling from the University of Portsmouth's School of Pharmacy and Biomedical Sciences, said, "The potential applications of this technique are far-reaching. The ability to tailor DNA origami structures with specific functionalities holds immense promise for advancing medical treatments and diagnostics.
"We envision a future where DNA origami structures could be used to deliver drugs or DNA directly to diseased cells, or to create highly sensitive diagnostic tools."
Current applications of DNA origami in biomedicine include vaccines, biological nanosensors, drug delivery , structural biology , and delivery vehicles for genetic materials.
Co-author Dr. Andrey Revyakin, formerly from the University of Leicester, said, "My lab has struggled for years to make DNA origami structures that remain functional in real-life biological applications. Dr. Rusling's triplex-based method, which 'upgrades' the classical DNA double-helix with an additional, third strand, stabilizes the DNA shapes, and does so with great precision, without affecting the functional modules of the molecule."
The paper says the new strategy is scalable and cost-effective, as it works with existing origami structures, does not require scaffold redesign, and can be achieved with just one DNA strand.
Dr. Rusling added, "What is really exciting about this technique is that it did not change the underlying origami DNA sequence, offering the ability to use these structures as carriers for synthetic genes."
Journal information: Journal of the American Chemical Society
Provided by University of Portsmouth
Explore further
Feedback to editors

Tiger beetles fight off bat attacks with ultrasonic mimicry

Machine learning model uncovers new drug design opportunities
5 hours ago

Astronomers find the biggest known batch of planet ingredients swirling around young star

How 'glowing' plants could help scientists predict flash drought

New GPS-based method can measure daily ice loss in Greenland

New candidate genes for human male infertility found by analyzing gorillas' unusual reproductive system
6 hours ago

Study uncovers technologies that could unveil energy-efficient information processing and sophisticated data security
7 hours ago

Scientists develop an affordable sensor for lead contamination

Chemists succeed in synthesizing a molecule first predicted 20 years ago

New optical tweezers can trap large and irregularly shaped particles
Relevant physicsforums posts, is it usual for vaccine injection site to hurt again during infection.
May 13, 2024
A Brief Biography of Dr Virgina Apgar, creator of the baby APGAR test
May 12, 2024
Who chooses official designations for individual dolphins, such as FB15, F153, F286?
The cass report (uk).
May 1, 2024
Is 5 milliamps at 240 volts dangerous?
Apr 29, 2024
Major Evolution in Action
Apr 22, 2024
More from Biology and Medical
Related Stories

Bridge in a box: Unlocking origami's power to produce load-bearing structures
Mar 18, 2024

Capturing DNA origami folding with a new dynamic model
Apr 18, 2024

New method can manipulate the shape and packing of DNA
Jun 16, 2023

Researchers devise genetically encoded DNA origami for targeted and precise gene therapy in vivo
Apr 25, 2023

DNA origami enables fabricating superconducting nanowires
Jan 19, 2021

Researchers craft 'origami DNA' to control virus assembly
Jul 17, 2023
Recommended for you

New gel breaks down alcohol in the body

Nanoparticle plant virus treatment shows promise in fighting metastatic cancers in mice

Research explores ways to mitigate the environmental toxicity of ubiquitous silver nanoparticles
May 11, 2024

High-speed atomic force microscopy helps explain role played by certain biomolecules in DNA wrapping dynamics
May 10, 2024

Designer peptoids mimic nature's helices

Nanoparticle researchers develop microfluidic platform for better delivery of gene therapy for lung disease
May 7, 2024
Let us know if there is a problem with our content
Use this form if you have come across a typo, inaccuracy or would like to send an edit request for the content on this page. For general inquiries, please use our contact form . For general feedback, use the public comments section below (please adhere to guidelines ).
Please select the most appropriate category to facilitate processing of your request
Thank you for taking time to provide your feedback to the editors.
Your feedback is important to us. However, we do not guarantee individual replies due to the high volume of messages.
E-mail the story
Your email address is used only to let the recipient know who sent the email. Neither your address nor the recipient's address will be used for any other purpose. The information you enter will appear in your e-mail message and is not retained by Phys.org in any form.
Newsletter sign up
Get weekly and/or daily updates delivered to your inbox. You can unsubscribe at any time and we'll never share your details to third parties.
More information Privacy policy
Donate and enjoy an ad-free experience
We keep our content available to everyone. Consider supporting Science X's mission by getting a premium account.
E-mail newsletter
- Alzheimer's disease & dementia
- Arthritis & Rheumatism
- Attention deficit disorders
- Autism spectrum disorders
- Biomedical technology
- Diseases, Conditions, Syndromes
- Endocrinology & Metabolism
- Gastroenterology
- Gerontology & Geriatrics
- Health informatics
- Inflammatory disorders
- Medical economics
- Medical research
- Medications
- Neuroscience
- Obstetrics & gynaecology
- Oncology & Cancer
- Ophthalmology
- Overweight & Obesity
- Parkinson's & Movement disorders
- Psychology & Psychiatry
- Radiology & Imaging
- Sleep disorders
- Sports medicine & Kinesiology
- Vaccination
- Breast cancer
- Cardiovascular disease
- Chronic obstructive pulmonary disease
- Colon cancer
- Coronary artery disease
- Heart attack
- Heart disease
- High blood pressure
- Kidney disease
- Lung cancer
- Multiple sclerosis
- Myocardial infarction
- Ovarian cancer
- Post traumatic stress disorder
- Rheumatoid arthritis
- Schizophrenia
- Skin cancer
- Type 2 diabetes
- Full List »
share this!
May 9, 2024
This article has been reviewed according to Science X's editorial process and policies . Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
Study finds association between neighborhood deprivation and DNA methylation in an autopsy cohort
by Impact Journals LLC
 and QQ plot (B) from the EWAS of DNAm with the ADI. Adjusted for race, sex, educational attainment, age at death, APOE genotype, cell type, and post-mortem interval. Bonferroni-threshold: 0.05/789889 = 6.33e-8 (λ=0.94). Credit: Aging (2024). DOI: 10.18632/aging.205764")
A new research paper was published in Aging , titled, "The association between neighborhood deprivation and DNA methylation in an autopsy cohort."
Previous research has found that living in a disadvantaged neighborhood is associated with poor health outcomes. Living in disadvantaged neighborhoods may alter inflammation and immune response in the body, which could be reflected in epigenetic mechanisms such as DNA methylation (DNAm).
In this new study, researchers from Emory University, University of British Columbia, BC Children's Hospital Research Institute, Centre for Molecular Medicine and Therapeutics, and Atlanta VA Medical Center used robust linear regression models to conduct an epigenome-wide association study examining the association between neighborhood deprivation (Area Deprivation Index; ADI), and DNAm in brain tissue from 159 donors enrolled in the Emory Goizueta Alzheimer's Disease Research Center (Georgia, U.S.).
"We found one CpG site (cg26514961, gene PLXNC1) significantly associated with ADI after controlling for covariates and multiple testing (p-value = 5.0e -8 )," write the researchers.
Effect modification by APOE ε4 was statistically significant for the top ten CpG sites from the EWAS of ADI, indicating that the observed associations between ADI and DNAm were mainly driven by donors who carried at least one APOE ε4 allele. Four of the top ten CpG sites showed a significant concordance between brain tissue and tissues that are easily accessible in living individuals (blood, buccal cells, saliva), including DNAm in cg26514961 (PLXNC1).
This study identified one CpG site (cg26514961, PLXNC1 gene) that was significantly associated with neighborhood deprivation in brain tissue . PLXNC1 is related to immune response , which may be one biological pathway to how neighborhood conditions affect health.
"The concordance between brain and other tissues for our top CpG sites could make them potential candidates for biomarkers in living individuals," the authors conclude.
Explore further
Feedback to editors

Tech alone can't replace human coaches in obesity treatment, study finds
2 hours ago

Research links sleep apnea severity during REM stage to verbal memory decline

Far from toxic, lactate rivals glucose as body's major fuel after a carbohydrate meal
4 hours ago

Study explores role of epigenetics, environment in differing Alzheimer's risk between Black and white communities

New gene therapy model offers hope for X-linked sideroblastic anemia treatment
5 hours ago

New tool allows for gene suppression in mouse heart muscle cells using CRISPRi

First study to globally map heat-wave-related mortality finds 153,000+ deaths associated with heat waves
6 hours ago

Body-wide molecular map explains why exercise is so good for you

Adiposity in childhood affects the risk of breast cancer by changing breast tissue composition, study suggests
7 hours ago

Study uncovers protein interactions as a potential path for ALS cure
Related stories.

Neighborhood deprivation tied to gestational diabetes risk
Feb 12, 2024

DNA methylation biomarker GrimAge version 2 described
Dec 21, 2022

International study reports the impact of genetics on epigenetic factors
Sep 9, 2021

Disadvantaged neighborhoods and symptoms of depression associated with premature aging
Jun 5, 2023

DNAmFitAge: Biological age indicator incorporating physical fitness
Jun 7, 2023

Exploring the effects of dasatinib, quercetin, and fisetin on DNA methylation clocks
Mar 6, 2024
Recommended for you

Stem cells provide new insight into genetic pathway of childhood cancer
8 hours ago

Congenital anomalies found to be ten times more frequent in children with neurodevelopmental disorders
Let us know if there is a problem with our content.
Use this form if you have come across a typo, inaccuracy or would like to send an edit request for the content on this page. For general inquiries, please use our contact form . For general feedback, use the public comments section below (please adhere to guidelines ).
Please select the most appropriate category to facilitate processing of your request
Thank you for taking time to provide your feedback to the editors.
Your feedback is important to us. However, we do not guarantee individual replies due to the high volume of messages.
E-mail the story
Your email address is used only to let the recipient know who sent the email. Neither your address nor the recipient's address will be used for any other purpose. The information you enter will appear in your e-mail message and is not retained by Medical Xpress in any form.
Newsletter sign up
Get weekly and/or daily updates delivered to your inbox. You can unsubscribe at any time and we'll never share your details to third parties.
More information Privacy policy
Donate and enjoy an ad-free experience
We keep our content available to everyone. Consider supporting Science X's mission by getting a premium account.
E-mail newsletter
To revisit this article, visit My Profile, then View saved stories .
- Backchannel
- Newsletters
- WIRED Insider
- WIRED Consulting
Will Knight
Google DeepMind’s Groundbreaking AI for Protein Structure Can Now Model DNA

Google spent much of the past year hustling to build its Gemini chatbot to counter ChatGPT , pitching it as a multifunctional AI assistant that can help with work tasks or the digital chores of personal life. More quietly, the company has been working to enhance a more specialized artificial intelligence tool that is already a must-have for some scientists.
AlphaFold , software developed by Google’s DeepMind AI unit to predict the 3D structure of proteins, has received a significant upgrade. It can now model other molecules of biological importance, including DNA, and the interactions between antibodies produced by the immune system and the molecules of disease organisms. DeepMind added those new capabilities to AlphaFold 3 in part through borrowing techniques from AI image generators.
“This is a big advance for us,” Demis Hassabis , CEO of Google DeepMind, told WIRED ahead of Wednesday’s publication of a paper on AlphaFold 3 in the science journal Nature . “This is exactly what you need for drug discovery: You need to see how a small molecule is going to bind to a drug, how strongly, and also what else it might bind to.”
AlphaFold 3 can model large molecules such as DNA and RNA, which carry genetic code, but also much smaller entities, including metal ions. It can predict with high accuracy how these different molecules will interact with one another, Google’s research paper claims.
The software was developed by Google DeepMind and Isomorphic labs, a sibling company under parent Alphabet working on AI for biotech that is also led by Hassabis. In January, Isomorphic Labs announced that it would work with Eli Lilly and Novartis on drug development.
AlphaFold 3 will be made available via the cloud for outside researchers to access for free, but DeepMind is not releasing the software as open source the way it did for earlier versions of AlphaFold. John Jumper, who leads the Google DeepMind team working on the software, says it could help provide a deeper understanding of how proteins interact and work with DNA inside the body. “How do proteins respond to DNA damage; how do they find, repair it?” Jumper says. “We can start to answer these questions.”
Understanding protein structures used to require painstaking work using electron microscopes and a technique called x-ray crystallography. Several years ago, academic research groups began testing whether deep learning , the technique at the heart of many recent AI advances, could predict the shape of proteins simply from their constituent amino acids, by learning from structures that had been experimentally verified.
In 2018, Google DeepMind revealed it was working on AI software called AlphaFold to accurately predict the shape of proteins. In 2020, AlphaFold 2 produced results accurate enough to set off a storm of excitement in molecular biology. A year later, the company released an open source version of AlphaFold for anyone to use, along with 350,000 predicted protein structures, including for almost every protein known to exist in the human body. In 2022 the company released more than 2 million protein structures.

Gabrielle Caplan

The latest AlphaFold’s ability to model different proteins was improved in part through an algorithm called a diffusion model that helps AI image generators like Dall-E and Midjourney create weird and sometimes photo-real imagery. The diffusion model inside AlphaFold 3 sharpens the molecular structures the software generates. The diffusion model is able to generate plausible protein structures based on patterns it picked up from analyzing a collection of verified protein structures, much as an image generator learns from real photographs how to render realistic-looking snapshots.
AlphaFold 3 is not perfect, though, and offers a color-coded confidence scale for its predictions. Areas of a protein structure colored blue indicate high confidence, while red areas show less certainty.
David Baker , a professor at the University of Washington who leads a group working on techniques for protein design, has competed with AlphaFold. In 2021, before DeepMind open sourced its creation, his team released an independent protein-structure prediction inspired by AlphaFold. His own lab recently released a diffusion model to help model a wider range of molecular structures, but he concedes that AlphaFold 3 is more capable. “The structure prediction performance of AlphaFold 3 is very impressive,” Baker says.
Baker adds that it is a shame that the source code for AlphaFold 3 has not been released to the scientific community.
Hassabis, who leads all of Alphabet’s AI initiatives, has long taken a special interest in the potential for AI to accelerate scientific research . But he says the latest techniques being developed for AlphaFold, a highly specialized AI system, could prove useful for building more general systems that aim to exceed human capabilities on many dimensions.
If AI programs like Google’s Gemini become a lot more capable over the next decade, he says, “you could imagine them using things like AlphaFold as tools, to achieve some other goal.”
You Might Also Like …
In your inbox: Will Knight's Fast Forward explores advances in AI
He emptied a crypto exchange onto a thumb drive —then disappeared
The real-time deepfake romance scams have arrived
Boomergasms are booming
Heading outdoors? Here are the best sleeping bags for every adventure

Amanda Hoover

Kate Knibbs

Paresh Dave

Louise Matsakis

Matt Burgess

Kathy Gilsinan

Lydia Morrish
Google DeepMind and Isomorphic Labs reveal AI able to predict large swathes of molecular biology

Alphabet’s Google DeepMind and its sister company Isomorphic Labs have created a new AI model that they say can help predict both the structure and interaction of most molecules involved in biological processes, including proteins, DNA, RNA, and some of chemicals used to create new medicines. The new model is a potentially giant leap for biological research. The companies are allowing researchers working on non-commerical projects to query the model for free through an internet-based interface.
Isomorphic Labs, which was spun out of Google DeepMind, has also begun using the system internally to speed its efforts to discover new drugs. The company currently has partnerships with Eli Lilly and Novartis aimed at developing multiple drugs, although the specifics of which diseases the companies are targeting has not been revealed. Proteins are the building blocks of life and their interactions with one another and with other molecules are the mechanism through which life’s processes happen. Being able to predict those interactions more accurately will help researchers advance science. by helping them understand the mechanism behind diseases, and, potentially, how to better treat and cure them. Called AlphaFold 3, the new AI software represents a major update and expansion of capabilities beyond Google DeepMind’s previous AlphaFold 2 system . Researchers from the companies published a paper on AlphaFold 3 today in the prestigious scientific journal Nature . Demis Hassabis, who serves as CEO of both Google DeepMind and Isomorphic, described the new model’s interaction predictions as “incredibly important for drug discovery.” John Jumper, the senior researcher who heads the protein structure team at Google DeepMind, described AlphaFold 3 as “an evolution of AlphaFold 2, but a really big one that opens up new avenues.” He also said he was excited to see what researchers would do with the new model, noting that AlphaFold 2 had already opened up new areas of biological research that he could never have imagined. AlphaFold 2 has been cited more than 20,000 times in other published scientific papers and has been used to work on drugs for malaria, cancer, and many other diseases.
AlphaFold 2 and 3
Debuted in late 2020, AlphaFold 2 solved a grand scientific challenge because it was able to accurately predict the structure of most proteins simply from their DNA sequence. The company later published the system’s predicted structures for all 200 million proteins with known DNA sequences and made them freely available to scientists in a massive database. Prior to this, only about 100,000 proteins had known structural information. Knowing the shape and structure of a protein is often a key part of understanding how it will function. But proteins do not work in isolation. And AlphaFold 2 was not designed to predict how proteins would interact with one another—although scientists soon found ways to modify AlphaFold 2 to make some of these predictions. Nor could AlphaFold 2 predict protein interactions with other kinds of molecules, such as DNA, RNA, ligands, and ions, that are found inside living things. It also could not predict the interaction of these other molecules with one another. AlphaFold 3 can. The system is not always accurate, but represents a major leap forward in performance. According to tests conducted by Google DeepMind and Isomorphic, AlphaFold 3 can accurately predict 76% of protein interactions with small molecules, compared to 52% for the previous best predictive software. It can predict 65% of DNA interactions compared to the next leading system, which only achieves 28%. And in protein to protein interactions, it can predict 62% accurately, more than doubling what AlphaFold 2 could do. Like AlphaFold 2, AlphaFold 3 also includes a confidence score alongside its predictions that give scientists some indication of whether they should trust the system’s output. This reduces the chance that the AI model will experience the sort of “hallucinations”—plausible but inaccurate outputs—that have plagued recent generative AI models. Jumper said that so far researchers have found these confidence scores to be highly correlated with whether the structural and interaction predictions are accurate. In other words, the system is not likely to be confidently wrong. There are a few classes of proteins where AlphaFold 3 is still not accurate. These include proteins that scientists consider “intrinsically disordered,” meaning they only assume a particular structure in the presence of another protein or molecule, perhaps changing their shape radically depending on circumstance, according to Max Jaderberg, the chief AI scientist at Isomorphic Labs.
Bioweapons worries
While many, including former Google DeepMind cofounder Mustafa Suleyman , who is now heading up a new consumer AI division at Microsoft , and Dario Amodei, the confounder and CEO of Google DeepMind rival Anthropic, have warned that rapid advances in AI may lead to the proliferation of bioweapons by radically lowering the knowledge barrier to creating deadly pathogens, Jumper said Google DeepMind and Isomorphic had consulted more than 50 experts in biosecurity, bioethics, and AI safety and concluded that the marginal risk AlphaFold 3 might present in terms of bioweapons creation was far outweighed by the system’s potential benefits to science, including advancing human understanding of disease and finding possible treatments.
The two companies are also only allowing access to the model through an internet service that allows outside researchers to prompt the system and receive a prediction, but does not give them access to the model itself or its underlying computer code. Unlike some efforts to create large language models (LLMs) for biology that can be prompted in natural language to produce a formula for a compound with particular properties, AlphaFold 3 still requires someone to have a fairly good understanding of biology to use it effectively. In addition, any suggested molecular structure it predicts would still need to be produced or isolated in a lab, a process that also requires relatively specialized knowledge. AlphaFold 3 uses a significantly different AI design than its predecessor AlphaFold 2. While both AI models are based around transformers, a kind of artificial neural network architecture pioneered by Google researchers in 2017, Jumper said the team working on the new system replaced entire “blocks” of the large transformer that powered AlphaFold 2.
AlphaFold 2 relied heavily on evolutionary information about the proteins for which it was trying to predict structures, while AlphaFold 3 leans on this evolutionary signal far less, using it only at the first step of its structure prediction. Instead, the new system devotes the majority of its components to working through the physical shape of the molecules it is making predictions about.
AlphaFold 3 also uses a diffusion model, similar to ones used for popular text-to-image generation models such as OpenAI’s DALL-E 3 or Midjourney, to learn how to puzzle out the precise atomic structures of molecules. Overall, despite covering far more substances than AlphaFold 2, AlphaFold 3 is a simpler design, with fewer separate components, than its predecessor.
Latest in Tech

Crewmember mistakenly caused the ship’s engine to stall hours before a blackout led to the Baltimore Bridge collapse

GM-owned Cruise reached a more than $8M settlement with pedestrian who was dragged by robo taxi

The juice isn’t worth the squeeze for many college majors, new report reveals: Lifetime earnings simply can’t keep up with the cost of degrees

OpenAI and Google lean in to AI personal assistants. Is this AI’s killer app?

Google unveils Project Astra chatbot tech and brings ‘AI overview’ to search for all U.S. users

AI will hit the labor market like a ‘tsunami,’ IMF chief warns. ‘We have very little time to get people ready for it’
Most popular.

The collapsed Baltimore bridge will be demolished soon, and the crew of the ship that’s trapped underneath will be onboard when the explosives go off

The housing crisis in the U.S. is flipped upside down in Japan, where each home that’s occupied could be next to an empty one by 2033

Meet the boomers who’d rather spend $100k to renovate their homes than risk the frozen housing market: ‘It would be too hard to purchase anything else’

Consumers were deprived of rare bourbons, including Pappy Van Winkle’s 23-year-old whiskey, by alcohol overseers

TV chef Gordon Ramsay spends an extra $7.6 million on staff as U.K. restaurant empire losses triple

Hedge fund billionaire Ken Griffin says college protests are the result of a ‘cultural revolution’ and Harvard should ’embrace our Western values’

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Genome Biol
Genetic impacts on DNA methylation: research findings and future perspectives
Sergio villicaña.
Department of Twin Research and Genetic Epidemiology, St. Thomas’ Hospital, King’s College London, 3rd Floor, South Wing, Block D, London, SE1 7EH UK
Jordana T. Bell
Associated data.
Multiple recent studies highlight that genetic variants can have strong impacts on a significant proportion of the human DNA methylome. Methylation quantitative trait loci, or meQTLs, allow for the exploration of biological mechanisms that underlie complex human phenotypes, with potential insights for human disease onset and progression. In this review, we summarize recent milestones in characterizing the human genetic basis of DNA methylation variation over the last decade, including heritability findings and genome-wide identification of meQTLs. We also discuss challenges in this field and future areas of research geared to generate insights into molecular processes underlying human complex traits.
Supplementary Information
The online version contains supplementary material available at (10.1186/s13059-021-02347-6).
Introduction
The complexity of the human genome lies not only in its composition of billions of base pairs, but also in the chemical modifications that make it interpretable to enzymes and other molecular factors, through epigenetic mechanisms. DNA methylation has been the most widely studied epigenetic mark since 1948 when it was first reported [ 1 ]. In humans, DNA methylation consists of the covalent addition of a methyl group to cytosine residues—predominantly at CpG sites—by a family of enzymes called DNA methyltransferases (DNMTs) [ 2 , 3 ]. DNA methylation plays an important role in multiple processes during human development and over the life course, such as the regulation of transcription [ 4 – 6 ], genomic imprinting [ 2 , 4 ], maintenance of X-chromosome inactivation [ 7 ], chromosomal maintenance, and genomic stability [ 8 ].
With advances in high-throughput molecular techniques our understanding of DNA methylation has greatly increased in the past few decades. Multiple methods have been developed for profiling DNA methylation patterns across the human genome. Currently, the gold standard is bisulfite conversion of DNA followed by deep sequencing or whole genome bisulfite sequencing (WGBS, Table 1 ). However, the most extensively used methylation profiling technologies are microarrays assessing DNA methylation at a proportion of the 28 million CpG sites in the genome. To date, Illumina bead-chip platforms have been most popular, where pre-designed probes target bisulfite-converted DNA, followed by hybridization, single-base extension, and its detection [ 9 ]. Early models included arrays such as the Infinium HumanMethylation27 BeadChip (27K), targeting around 27,000 sites (0.1% of total CpGs) mainly in CpG islands (CGIs) within promoters [ 9 ], followed by the widely used Infinium HumanMethylation450 array (450K), targeting ∼480,000 sites (1.7% of total CpGs) consisting of the 27K sites and increased coverage in non-CGIs and intergenic regions [ 10 ]. A more recent version is the Infinium MethylationEPIC BeadChip (EPIC), targeting ∼850,000 sites (3% of total CpGs), which include almost all of the 450K sites, with additional CpG sites in enhancers [ 11 ].
Glossary of commonly used terms
Unlike DNA sequence, genomic methylation patterns are not directly inherited during meiosis [ 12 ], but are mostly reprogrammed in two waves during embryogenesis [ 13 – 15 ]. Following this, DNA methylation modifications can be both stable and dynamic during mitosis events that accumulate over the life course [ 16 , 17 ]. These observations suggest that the environment may be a key driving force behind changes in mitotic DNA methylation [ 17 – 20 ]. However, growing evidence now shows that genetic variation also plays a role in the establishment of DNA methylation marks, independently of or in contribution with environmental exposures.
Research interest in genetic impacts on DNA methylation variation is especially relevant in context of methylome changes observed in disease [ 16 , 21 – 23 ], alongside results from genome-wide association studies (GWASs). Although many genetic associations have been identified from GWASs, there remain important unanswered questions about candidate causal variants and their functional consequences, as GWAS signals tend to fall in non-coding regions [ 24 ]. Methylome analyses can provide a valuable piece of information as a post-GWAS resource, giving insights into regulatory genomic potential of GWAS signals and helping to prioritize loci to further follow-up [ 25 – 27 ].
Given these considerations, here, we present an overview of results identifying genetic drivers of DNA methylation variation. We discuss methylation heritability findings, and then focus on genome-wide studies that have identified genetic variants as meQTLs for DNA methylation profiles. We also discuss cellular mechanisms that may explain genetic impacts on DNA methylation levels. Lastly, we consider challenges of meQTL analyses, as well as novel applications and further research directions.
DNA methylation heritability
A fundamental question in the study of human traits is to assess the extent to which a phenotype is under the influence of genetic factors, that is, how heritable it is. Heritability refers to the proportion of phenotypic variance attributed to either total genetic effects (broad-sense heritability, H 2 ), or additive genetic effects (narrow-sense heritability, h 2 ) [ 28 ], where the latter is most commonly estimated in context of DNA methylation analyses.
For the estimation of DNA methylation heritability, most studies apply twin-based study designs. The underlying premise of the twin design is based on trait comparison between monozygotic (MZ) twin pairs who share typically 100% of their genome variation, compared to dizygotic (DZ) twins who share on average only 50% of genetic variation. The narrow-sense heritability is then calculated by comparing the correlation of a trait—here level of DNA methylation at a genomic region—between MZ and DZ twins [ 29 ], following a series of assumptions. In a recent study in whole blood samples from 2603 individuals from the Netherlands Twin Registry, van Dongen et al. [ 30 ] estimated the individual CpG site heritability to range from 0 to 0.99 at each CpG site profiled on the Illumina 450K array, where the mean genome-wide heritability averaged over all CpG sites tested was h 2 ¯ ^ = 0.19 ( h 2 ¯ ^ = 0.20 with the classical twin method). The estimate of the average CpG site heritability across the methylome as 0.19 is in agreement with previous twin methylation heritability studies using the 450K [ 31 ] and 27K arrays [ 32 ]. Furthermore, the study estimated that approximately 41% of Illumina 450K sites had significant evidence for additive genetic effects and suggested that heritability at a proportion of DNA methylation sites is sex- and age-specific.
Fewer studies estimate DNA methylation heritability using other approaches, for example, using familial clustering models in extended families. The advantage of such methods is their wider applicability to multiple types of relatives beyond twins and circumventing key assumptions of the classical twin model such as equal influence of common environment for MZ and DZ twins and independence of genetic and environmental factors. Despite this, DNA methylation heritability estimates from familial clustering studies are consistent with those obtained from classical twin models. McRae et al. [ 33 ] estimated the heritability of DNA methylation measured using the Illumina 450K array in 614 peripheral blood leukocyte samples from twin pairs, their siblings and fathers, in altogether 117 families of European descent from the Brisbane System Genetics Study. The estimates of heritability across the Illumina 450K probes give a similar mean CpG site genome-wide estimate of h 2 ¯ ^ = 0.187 , ignoring probes with known genetic variants ( h 2 ¯ ^ = 0.199 , if all probes included). Using a different approach, Nustad et al. [ 34 ] designed a Bayesian mixed model that could include pedigree structure for estimating heritability in two sets of CD4 + T cell samples ( n =995 and n =530) from the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study, profiled on the 450K array. Here, the mean heritability point estimates across the genome ( h 2 ¯ ^ = 0.33 and h 2 ¯ ^ = 0.36 ) are slightly higher compared to other studies, potentially because the mean was calculated only considering CpG sites with strong evidence for non-zero heritability, as well as lack of precise estimates of shared environmental effects. Other studies using the 450K array have found comparable average heritability estimates based on family clustering ( h 2 ¯ ^ = 0.09 [ 35 ] and H 2 ¯ ^ = 0.13 [ 36 ]), or other methods applicable to unrelated individuals such as SNP-based heritability, calculated using all genetic variants [ 30 , 37 ]. For instance, in 3948 blood samples from the Avon Longitudinal Study of Parents and Children (ALSPAC), Gaunt et al. [ 37 ] estimated the genome-wide average SNP-based heritability for 450K array probes in blood at different time points over the life course to range between 0.20 and 0.25, based on a panel of 1.2 million common SNPs. The majority of methylation variance was explained by SNPs located over 1 Mbp away from the methylation site (or in trans ).
Overall, these heritability studies indicate that DNA methylation profiles have a genetic basis, which expressed as the average heritability across all CpG site in the genome profiled by the Illumina 450K array, ranges from 0.1 to over 0.3. Although this genome-wide mean estimate of methylation heritability could be considered moderate or low, the heritability distribution at specific CpG sites ranges from 0 to 1, and at least one tenth of profiled sites are highly heritable ( h 2 ^ > 0.5 ) [ 30 , 31 , 33 ]. Furthermore, because genetic variability differs across populations and over time, heritability estimates are population- and age-specific, which may explain some of the differences in reported mean DNA methylation heritability estimates so far [ 28 ]. Another factor to consider when interpreting the heritability estimates is that they may vary according to DNA methylation platform [ 38 , 39 ], as array technologies only cover a limited proportion of CpGs out of the 28 million CpGs genome-wide (approximately 1.7% for 450K, 3% for EPIC) and regulatory elements tend to be underrepresented (see the “ DNA methylation profiling ” section).
An outstanding research question has considered evidence for transgenerational transmission of DNA methylation patterns independent of genetic variation or transgenerational epigenetic inheritance. In model organisms such as mice and rats, several phenotypes have been linked to DNA methylation transgenerational inheritance. Examples include a kinked tail phenotype caused by methylation in a retrotransposon within the axin-fused allele in mice [ 40 ], and metabolic phenotypes in male rats linked to in utero nutritional deficiencies and alterations in the sperm methylome [ 41 ]. In contrast, human transgenerational epigenetic inheritance studies are limited and show negative results, suggesting that genetic variants likely fully explain the observed methylation heritability. In a study aiming to test whether methylation levels at certain CpG sites are inherited in a Mendelian fashion through multiple generations in 16 families (123 subjects) from the Arab population, Zaghlool et al. [ 42 ] inspected loci where blood DNA methylation levels followed a trimodal distribution, that is, with peaks around 0 (unmethylated), 0.5 (hemi-methylated), or 1 (methylated). Although about a thousand CpG sites from the 450K followed such patterns, in almost all cases, DNA methylation changes were associated with nearby genetic variants (within 1 Mbp or less), discarding a direct mechanism of transmission that is independent of genetic variation. Importantly, the trimodal loci had high mean heritable values (0.8±0.18), and almost half were associated with expression quantitative trait loci (eQTLs). McRae et al. [ 33 ] reached similar conclusions, noting that the transgenerational inheritance of DNA methylation is mainly attributable to genetic heritability. Therefore, so far, there is no robust evidence in humans to indicate that DNA methylation heritability may be attributed to non-genetic effects, such as evidence for transgenerational epigenetic inheritance as reported in other species [ 12 ].
Methylation quantitative trait loci
Given the observed evidence for DNA methylation heritability, much interest has focused on identifying specific genetic variants that influence DNA methylation variation across the genome. Multiple studies have explored the correlation between DNA methylation levels and genetic variants across the genome (typically single nucleotide polymorphisms, SNPs), to identify DNA methylation quantitative trait loci or meQTLs (also referred to as mQTLs or metQTLs). Although several early papers tackled meQTLs identification over limited target sites [ 43 – 46 ], it was not until the early 2010’s that initial genome-wide efforts identified meQTLs on the 27K methylome and across multiple tissues (Gibbs et al. [ 47 ], Zhang et al. [ 48 ] and Bell et al. [ 49 ]).
Studies to date have reported an influence of meQTLs on up to 45% of CpG sites profiled by the Illumina 450K array across the genome [ 31 , 35 , 50 , 51 ], with more than 90% of meQTLs acting on nearby methylation sites (in cis ) [ 38 , 50 , 52 ]. CpG sites that have higher heritability estimates are more likely to be associated with meQTLs in cis , trans , or both, and have a clear polygenic architecture [ 35 , 38 , 50 ]. Some studies also include replication in independent sample sets, although overall a direct comparison of meQTL signals can be challenging because studies do not systematically report meQTL effect sizes. Despite observations that meQTLs tend to have moderate to large effects, the “missing heritability” issue has also been raised in the context of meQTLs. That is, family-based heritability estimates of DNA methylation are greater than the proportion of variance explained by meQTLs, especially for distal associations [ 37 , 53 – 56 ].
Detecting meQTLs
MeQTL identification is based on association tests between genetic variation genome-wide and DNA methylation levels at a specific CpG site (Fig. 1 ). As for other quantitative trait analyses, the majority of meQTL detection approaches apply linear models, where the DNA methylation level at a CpG site is the response variable and genetic variants are predictors along with technical and biological covariates, such as smoking and age. Other statistical tests employed include non-parametric methods such as Spearman rank correlation [ 51 , 57 – 59 ] and Kruskal-Wallis rank test [ 60 ], that do not make assumptions about the distribution of variables, or even machine learning approaches such as random forests [ 61 ].

A typical workflow for meQTL identification. Step 1 is DNA methylation profiling. The most commonly applied methylation profiling technologies in meQTL studies are Illumina methylation arrays and whole genome bisulfite sequencing (WGBS). In both approaches, DNA is treated with bisulfite, converting unmethylated cytosines into uracils, and leaving methylated cytosines unchanged. DNA can then be profiled by sequencing or by Illumina array technologies, consisting of pre-designed probes. In step 2 , DNA methylation levels at each CpG site are quantified, typically either as percentage (0–100%, e.g., in WGBS) or proportion methylation (0–1, e.g., in the Illumina technology methylation β -value). The example shows the distribution of methylation β -values for one CpG site ( m 1 ) across all profiled samples. Step 3 is the association of a set of genetic variants (coded as allele dosages at each locus) with methylation values at each CpG site, usually using linear models. In this example, after the association test at site m 1 with a set of i genetic variants (shown in the Manhattan plot), g 1 was found to be significantly associated with m 1 (shown in the boxplot). Finally, step 4 represents the extension of the genetic association test to all profiled CpG sites genome-wide and the identification of genome-wide meQTLs after setting an appropriate threshold for statistical significance. The resulting meQTL associations can be either short-range, in cis (shown in heatmap for a few Mbp), or long-range or on different chromosomes, in trans (shown in Circos plot with all chromosomes)
In most studies the focus is on detecting evidence for additive genetic effects alone, where the genetic predictor is the dose of the alternative allele, for example, 0 for genotype “AA”, 1 for “Aa” and 2 for genotype “aa”. To date and to our knowledge, full genome-wide meQTL analyses have not yet considered genetic association models including dominance effects or overall genotype effects. However, Zeng et al. [ 62 ] explored meQTLs at 984 CpGs with parent-of-origin effects (POE) in 5101 individuals from Scottish families. The model included additive effects (coded as the dosage of alternative allele), dominance effects (coded 1 for heterozygotes and 0 for homozygotes), and POE effects (coded 0, −1, and 1 for homozygotes, “Aa” and “aA”, respectively). Likewise, some studies focusing on subsets of CpGs have identified meQTLs in gene interaction models, specifically gene-by-gene (G ×G) and gene-by-environment (G ×E) (see the “ Gene–environment interactions ” section).
The majority of studies discussed here apply Illumina DNA methylation arrays. In these platforms, the DNA methylation level at a CpG site is quantified through the Illumina methylation β -value, defined as the intensity measured in the methylated probes for that CpG over the total intensity across all probes for the CpG and a constant. The methylation β -value is often interpreted as the probability of methylation at a given site, or the proportion of methylated cells in the sample (Fig. 1 ). Some studies apply transformations of methylation β -values—such as the logit transformation or M -value—which are more appropriate to control for heteroskedasticity but are perhaps less biologically interpretable [ 63 , 64 ].
In addition to meQTL studies that explore DNA methylation levels using Illumina arrays, several meQTL approaches have also applied sequencing technologies. To date, only one study has used sequencing techniques to detect meQTLs across the full genome, rather than focusing on specific genomic subsets. In a sample of 697 Swedish subjects, McClay et al. [ 52 ] used methyl-CpG-binding domain (MBD)-enriched sequencing (MBS-seq) genome-wide and profiled ∼13M CpGs collapsed into 4.5M loci across the genome. DNA methylation was quantified by estimating the coverage at each CpG. The results show that 15% of methylation loci have meQTLs (primarily within 1 Mbp), and 98% of the tested SNPs were associated with at least one CpG. Other studies have employed strategies such as targeted bisulfite sequencing of a pre-designed panel with informative genomic regions [ 65 ], MeDIP-sequencing at candidate regions [ 66 ], and meQTL replication in WGBS data [ 67 ]. Several studies have also explored sequence-dependent allele-specific methylation (ASM), which represents a specific type of meQTL effect in cis . In contrast to meQTL analysis, ASM discovery is restricted to heterozygous regions within single samples, and comparison of differentially methylated CpG sites (DMSs) between the two distinct alleles, for example, using Fisher’s exact test or equivalent. ASM studies to date have been carried out using bisulfite sequencing in a moderate number of samples (less than 100). ASM results show that around 10% of the explored CpGs exhibit allelic imbalance at heterozygous regions [ 65 , 68 – 70 ], which is consistent with meQTLs results.
Distribution of meQTLs across the genome
Meqtls can have local or distal effects.
MeQTLs can be divided into two classes based on the proximity of the genetic variant to the CpG site. Cis -meQTLs are genetic variants near to or proximal to the target CpG site, and trans -meQTLs are separated by one or more Mbp from the target CpG or located on different chromosomes. Identification of cis and trans -meQTLs includes testing for associations across all possible pairs of SNPs-CpGs. Pairs can be categorized into “proximal” or “distal” and multiple testing correction can be applied for each group independently, or they can be analyzed together and annotated post hoc [ 38 , 47 , 71 ]. Correcting for multiple testing burden is a crucial step for the definition of genome-wide significant p -value thresholds. Published thresholds are typically of the order of p <1×10 −5 for cis effects and p <1×10 −9 for trans effects, based on applying permutation-based approaches to estimate the false discovery rate (FDR) or Bonferroni correction to control the family-wise error rate (FWER). The exact multiple testing correction threshold clearly depends on the methylation array and genotype coverage, methylation, and genotype distributions, as well as sample structure and sample size if permutation-based approaches are applied (see the “ Multiple-testing correction ” section). Some studies limit the search to cis -meQTLs alone, reducing the number of total tests, or carry out trans associations only for selected SNP-CpG pairs [ 51 , 60 ].
To date, the primary focus has been on cis -meQTLs identification. In general, studies with large sample sizes (> 1000) have estimated that at least 10% [ 38 , 53 – 55 ] and up to 45% [ 35 , 50 , 51 ] of the methylome is influenced by nearby meQTLs. A consideration in cis -meQTL analysis protocols is the maximum distance between genetic variants and CpG sites. Published studies have applied a range from a few kbp to 1 Mbp, but in almost all cases it has been observed that the strength of the cis -meQTL effect is inversely proportional to the distance between genetic variant and CpG site. For example, in one of the early genome-wide meQTL analysis using 27K DNA methylation levels in lymphoblastoid cell lines (LCLs), 37 CpG sites had meQTLs in genome-wide analyses across all possible SNP-CpG pairs, but for 27 of these sites the most significant meQTL was located within 50 kbp of the CpG site [ 49 ]. More recently, Hannon et al. [ 38 ] conducted a genome-wide analysis across all SNPs-CpGs using EPIC DNA methylation levels in 1111 blood samples. The results identified meQTLs at 12% of assayed methylation sites, and again a predominance of these associations occurred in cis . Higher effect sizes were observed for genetic variants within a maximum of 500 kbp from the CpG site (in cis ), where the average of the change in DNA methylation per allele was of 3.48%, compared to 3.26% in trans .
Conversely, meQTL genome-wide association analyses to date agree that no more than 5% of total CpGs show evidence for trans -meQTLs. The exception to this are the results from Gong et al. [ 72 ] estimating meQTLs in different cancer tissues samples. The observation of higher trans -meQTL proportions here (more than 10% of total CpG sites are associated with trans -meQTLs in eight cancer types) suggests that under certain conditions, the effects of the distal associations could be enhanced. Furthermore, although trans -meQTL are relatively rare genome-wide, these effects also tend to target specific genomic regions (see the “ MeQTLs are differentially distributed across the genome ” section).
The physical threshold for categorizing a meQTL as cis or trans matters. Insights into the distance between cis -meQTLs and the target CpG sites were gained by Banovich et al. [ 67 ] who used a relatively small cis window of 6 kbp to detect meQTLs in LCLs. The authors estimated the median distance of putative causal cis associations as 76 bp, with 87% of the meQTLs located within 3 kbp of the CpGs. At the other end of this spectrum, Huan et al. [ 35 ] report that 70% of intra-chromosomal trans -meQTLs were within 5 Mbp of the target CpG, leading to the conclusion that such associations may act as long-range cis -meQTLs, rather than as trans . In contrast, inter-chromosomal associations are the most commonly reported trans -meQTLs, accounting for at least 65% of the trans -meQTLs [ 35 , 37 , 50 , 51 , 55 ]. Another factor to take into account is that some trans associations could be SNPs in long-range linkage disequilibrium (LD) with “real” cis -meQTLs—as observed for 17% of intra-chromosomal SNP-CpG associations in lung tissue, after conditional analysis [ 56 ].
MeQTLs are differentially distributed across the genome
Early efforts exploring the correlation between genetic variants and DNA methylation showed evidence that meQTLs and their target DNA methylation sites are not randomly distributed in the genome. Non-genic regions and enhancers appear to be hotspots for CpG sites associated with cis -meQTLs, while CpG islands (CGIs), 5 ′ untranslated regions (UTRs) and regions upstream of the transcription start sites (TSSs) show depletion of CpG sites with cis -meQTLs. In contrast, the opposite pattern is observed for CpGs with trans -meQTLs, which are enriched in CGIs and in promoters and regions surrounding the TSSs, and are underrepresented in gene bodies, 3 ′ UTRs, and heterochromatin regions [ 35 , 37 , 50 , 51 , 53 , 56 , 67 , 73 ]. This genomic distribution of meQTL-related CpGs appears to be quite stable during several life stages [ 37 ] and across tissues [ 73 ].
The underrepresentation of CpGs with cis -meQTLs in CGIs is related to the observation that most of the tested CpGs in CGIs fall in gene promoters, where they tend to be constitutively hypomethylated and have lower DNA methylation variances [ 5 , 6 ]. As hypothesized by Do et al. [ 70 ], meQTL-associated CpGs may be located in areas with more flexible evolutionary constrains, in contrast to typically hypomethylated CGIs which are conserved across vertebrate promoters [ 74 ]. This hypothesis is also supported by results from Husquin et al. [ 60 ] who observed that DMSs in monocytes between two populations (78 samples of African descent and 78 of European descent) are enriched to harbor cis -meQTLs (70.2% of DMSs have cis -meQTLs) compared to the genome-wide meQTL proportion (12.6% of EPIC sites had cis -meQTLs). Hence, CpGs, where methylation patterns are less conserved across different populations, have a higher probability of being under the influence of meQTLs.
The genetic variants driving meQTL effects also exhibit non-random genomic distributions. Min et al. [ 50 ] found that active chromatin domains and genic regions were enriched for meQTLs that act in cis only or both in cis and trans , while heterochromatin and intergenic regions were enriched for trans only meQTLs. Using a different approach, an analysis at 11.5 million DNA methylation sites profiled by WGBS in 34 samples [ 75 ] identified 221 de novo DNA motifs associated with unmethylated regions, and 92 motifs associated with methylated regions. Using data from previously published studies, the authors found that DNA motifs associated with methylation were enriched in meQTLs variants, especially near TSSs.
Lastly, trans -meQTLs results show that the number of inter-chromosomal trans -meQTLs is usually proportional to the number of genes in a chromosome, except for chromosomes 16 and 19 which are highly enriched for trans -meQTLs and chromosome 1 which is depleted for trans -meQTLs [ 37 , 53 ]. Also, McRae et al. [ 53 ] estimated that almost 25% of trans -meQTLs are located in telomeres and sub-telomeres. The major histocompatibility complex (MHC) region is another locus that harbors highly heritable CpGs and meQTLs associated with multiple CpGs [ 33 , 76 , 77 ].
Tissue-specificity of meQTL effects
DNA methylation plays an important role in cell lineage and tissue differentiation, resulting in tissue-specific methylation profiles over a considerable proportion of the methylome. Most meQTL studies explore whole blood, but analyses within specific cell types or bulk tissue have also been carried out.
MeQTLs in blood-based samples
Most studies have identified meQTLs in blood and blood-derived cells, including whole blood, LCLs, peripheral blood mononuclear cells (PBMC), and leukocytes (see Table 2 ). Blood-based meQTLs studies are most common to date, have larger sample sizes, and have shown high replicability. The majority of blood reports are not limited to the discovery of novel meQTLs alone, but also include study designs that integrate DNA methylation findings with GWAS results or other biological data. In the largest study to date, the Genetics of DNA Methylation Consortium (GoDMC), a multi-cohort meta-analysis meQTL resource, combined data from 32,851 blood samples across different population cohorts and found that 45.2% of CpGs in the 450K array have meQTLs, with greater effect sizes for cis associations [ 50 ]. Additionally, the authors detected substantial sharing between meQTLs and GWAS signals, and constructed a network of CpG sites that share meQTLs, identifying 405 highly interconnected genomic communities enriched for regulatory genomic features and links to complex traits. Huan et al. [ 35 ] performed an analysis in 4170 whole blood samples, identifying 4.7 million cis -meQTLs (within 1 Mbp of target CpG) and 706 thousand trans -meQTLs. After a follow-up analysis, the authors found 92 CpGs with a likely causal role in cardiovascular disease, as well as supporting evidence of CpG-expression contribution to these putative causal pathways. Likewise, Bonder et al. [ 51 ] studied trans -meQTLs focusing on SNPs previously associated with complex traits. Using 3841 whole blood samples from the Netherlands, they showed that one-third of the analyzed SNPs affect DNA methylation levels at 10,141 CpG sites in trans , and where 95% of trans -meQTLs were validated in external data from 1748 lymphocytes. Furthermore, the authors provided several examples of trans -meQTLs with effects on specific transcription factors levels as well as methylation of their binding sites across the genome. Chen et al. [ 78 ] identified cis -meQTLs in immune cells (CD14 + monocytes, CD16 + neutrophils, and naive CD4 + T cells) at almost 10% of the CpG sites from the Illumina 450K, and estimated relatively low blood cell specificity of meQTLs especially between myeloid cells.
Blood-based genome-wide meQTL studies (sample size > 100) in whole blood or blood-derived cell samples
a If not specified, the sample type is whole blood. If more than one sample per analysis, the pooled size and number of samples is reported
b In parenthesis, maximum or minimum distances are indicated for cis and trans analysis, respectively. The range of results is presented if more than one analysis was done (unless otherwise stated)
c Multiple-testing criteria, with the corresponding p -value threshold for cis and trans meQTLs (where it differs). Different approaches to estimate FWER and FDR are as follows:
1 FWER based on Bonferroni correction
2 FWER based on Holm-Bonferroni correction
3 FDR based on permutations
4 FDR based on Benjamini-Hochberg correction
d Reference panel for imputations
e Database or biobank
FWER family-wise error rate, FDR false discovery rate, LCL lymphoblastoid cell lines, WGS whole genome sequencing, MCC-seq methylC-capture sequencing, WGBS whole genome bisulfite sequencing, MBD-seq methyl-CpG-binding domain sequencing, 1000G 1000 genotypes, GoNL Genome of the Netherlands, TF transcription factor, ASM allele-specific methylation
MeQTLs in non-blood-based tissues and cells
Genome-wide meQTLs have also been identified in a range of tissues including several regions of the brain, lung, skeletal muscle, buccal and saliva samples, placenta, and adipose tissue (Table 3 ). The discovery of meQTLs across brain regions [ 57 , 71 , 79 , 80 ], their overlap with non-brain tissue findings [ 70 , 73 ] (see the “ Tissue-shared meQTLs-CpGs ” section), and their co-localization with other molecular QTLs [ 81 ] has initiated further studies to identify and characterize the role of genetic variants underlying neurological disorders. In lung tissue, Morrow et al. [ 82 ] investigated meQTLs that may impact the pathogenesis of chronic obstructive pulmonary disease in 90 cases and 36 controls. The authors found cis -meQTLs at 10% of the 450K CpGs, and significant overlaps with GWAS signals for the disease. In parallel, Taylor et al. [ 83 ] assessed 282 samples of skeletal muscle on the Illumina EPIC array and found cis -meQTLs for almost 21% of CpGs. In adipose tissue, Grundberg et al. [ 31 ] ( n =603, from UK females) and Volkov et al. [ 76 ] ( n =119, from Scandinavian males) identified the cis and trans genetic effects on the methylome profiled by the 450K array. Both studies identified meQTLs that may also be involved in metabolic traits, such as variants in the ADCY3 gene, associated with obesity and BMI.
Overview of published genome-wide DNA methylation quantitative trait loci studies in blood-independent sample types
a We account for the different association analyses, even if they are published in the same paper
b If more than one analyses is available, the range is presented
Tissue-shared meQTLs-CpGs
The majority of DNA methylation signatures are tissue-specific and reflect the developmental trajectories of each cell line [ 13 ]. However, when DNA methylation levels are partially or fully driven by genetic variants, DNA methylation levels and meQTLs effects can be tissue-specific or they can also be shared across tissues. Several studies have explored this question, focusing on how easily accessible tissues such as blood may be used as proxies for the indirect study of difficult-to-reach tissues. In a report including samples from T cells, temporal cortex, neurons, glia, and placenta profiled with the 450K array, Do et al. [ 70 ] found good overlap in the percentage of meQTL-associated CpGs between temporal cortex with those in neurons/glia (61%) as expected, but not with T cells (28%) or placenta (12%). However, the study explored in a small to moderate sample size ( n ≤54 for each sample type), and consequently had limited power for detection of modest effects and their tissue-specificity assessment. Lin et al. [ 73 ] explored meQTLs in 197 saliva samples from control and schizophrenia/schizoaffective disorder patients and compared their results with two previous studies in brain and blood samples. They estimated that 38–73% of the meQTL variants in each tissue are shared with another and that most have a consistent effect direction across tissues. They found that 31–68% of the significant CpGs harboring meQTLs in a certain tissue are also significant in at least one other tissue. From these results, the tissues that share most meQTLs or most CpGs with meQTLs—with at least one other tissue—were blood and saliva. Another interesting observation was that tissue-shared signals were enriched in genetic risk loci of diseases such as schizophrenia, as well as in cross-tissue eQTLs (i.e., eQTLs significant in both blood and brain tissue). Similarly, Qi et al. [ 84 ] assessed the correlation of genetic effects at the peak cis -meQTLs in blood and brain from five data sets profiled on the Illumina 450K array. The correlation of meQTL effects between two sets of samples profiled in the same tissue was strong (correlation coefficient r b ^ = 0.92 for both blood and brain sample types), and lower, although still considerable, between brain and blood samples ( r b ^ = 0.78 ). Other cross-tissue meQTL analyses have also included comparisons between blood, brain, adipose tissue, breast, kidney, and lung samples [ 50 , 56 , 65 , 85 , 86 ].
Although no clear consensus currently exists in the estimated proportion of tissue-shared meQTLs, increasing evidence shows that a major subset of meQTL-CpG pairs are indeed shared among multiple tissues and cell types.
MeQTLs databases
Several efforts have attempted to create databases of meQTL findings. One of the first online repositories that incorporated results from GWAS of DNA methylation was GRASP, where the current build has 52,419 meQTLs records [ 87 , 88 ]. In 2015, Relton et al. [ 89 ] constructed the Accessible Resource for Integrated Epigenomic Studies (ARIES), summarizing findings from DNA methylation analysis of 1018 mother-offspring pairs from the Avon Longitudinal Study of Parents and Children (ALSPAC). The resource also includes one of the few longitudinal meQTL studies to date, complementing the original database [ 37 ]. The Brain xQTL Serve is another resource that reports results of genetic variation in three molecular traits—gene expression, DNA methylation, and histone acetylation—from prefrontal cortex samples of two longitudinal aging cohorts [ 57 ]. In cancer research, the Pancan-meQTL [ 72 ] and DNMIVD (for DNA Methylation Interactive Visualization Database) [ 90 ] use data from The Cancer Genome Atlas (TCGA). Pancan-meQTL reports 8028 cis and trans -meQTLs identified in 7242 samples from 23 different tumor types, while DNMIVD complements the Pancan-meQTL findings with additional analyses, such as diagnostic and prognostic models, and pathway-meQTL. Hannon et al. [ 38 ] published an interactive database of meQTLs from a blood-based study in 1111 samples, along with putative pleiotropic associations of meQTLs and multiple traits. Altogether, QTLbase is probably the most comprehensive resource to date in different sample types. It compiles summary statistics for molecular QTLs from 233 studies, with meQTL associations representing 16% of the database and summarizing results from 39 meQTLs publications in different tissue types [ 91 ]. In blood specifically, the GoDMC resource [ 50 ] includes an online searchable tool with a full list of meQTLs from the largest blood meQTL study to date (see the “ MeQTLs in blood-based samples ” section).
Genetic effects on DNA methylation: potential underlying mechanisms
Cis -meqtl mechanisms.
Despite the identification of hundreds of thousands of associations between meQTLs and CpGs, the molecular mechanisms underlying meQTLs are not well characterized. The leading hypothesis to explain cis -meQTL effects is that SNPs in protein binding sites alter or disrupt the activity of sequence-specific binding proteins—such as transcription factors (TFs)—and change methylation patterns of nearby CpGs, either directly or through a signaling cascade [ 59 , 67 , 70 , 92 , 93 ]. In support of this hypothesis, Banovich et al. [ 67 ] showed that for meQTLs in TF binding sites (TFBSs), different alleles predicted to affect affinity of TF binding were correlated with methylation levels at nearby CpG sites. Wang et al. [ 75 ] also showed consistent findings by identifying DNA motifs associated with methylation levels, as previously described (see the “ MeQTLs are differentially distributed across the genome ” section). The authors profiled binding profiles of 845 TFs and concluded that TFs can interact with DNA motifs that are also associated with DNA methylation levels. These results are also in concordance with mechanisms reported to underlie other DNA regulatory pathways and their QTLs, such as histone modifications and RNA polymerase II [ 94 ].
The signaling pathways triggered by sequence-specific binding proteins are still under discussion, but the main premise is that if a TFBS is occupied, this could be enough to prevent DNA methylation changes in the vicinity of this TFBS. This would represent a form of passive control of genetic variation on DNA methylation, via TFBS occupancy (Fig. 2 a). Alternatively, TFs could recruit DNMT3A and TET enzymes for active methylation or demethylation (Fig. 2 b). This is supported by the observation of an overlap of TFBS with methylation-associated DNA sequence motifs [ 75 ].

Mechanisms underlying cis -meQTL effects. a Passive mechanism. Under normal conditions a sequence-specific binding protein (such as CTCF) can bind to its target and prevent methylation changes at surrounding CpG sites due to its occupancy. If a meQTL disrupts the site, the protein cannot bind successfully, and the CpG sites are prone to change in baseline methylation status. b Active mechanism. If a meQTL is located in a TFBS, lack of TF binding can promote the recruitment of DNMT or TET enzymes, and thus modify the methylation status of nearby CpG sites
One of the main examples in support of the hypothesis of passive genetic control on DNA methylation is CTCF (CCCTC-binding factor), which is an insulator involved in chromatin regulation, forming loops and bringing together genetic elements that may be physically far apart. CTCF binding sites usually contain CGI motifs and have to be poorly methylated to allow for the recruitment of the protein [ 95 ]. The occurrence of a meQTL within the CTCF binding site may result in a decrease or even annulment of CTCF binding affinity, which in turn can lead to an increase in DNA methylation of nearby CpG sites, as shown in the mouse methylome [ 96 ]. Multiple studies have now highlighted CTCF binding as a key example of cis genetic-epigenetic interactions [ 56 , 70 , 71 , 79 , 97 ].
An example of cis -meQTL active mechanisms involves a genetic variant within the gene underlying a clinical subgroup of colorectal cancer known as MSI+ (or microsatellite-unstable cancer). Here, decrease of gene expression of the DNA mismatch repair gene MutL homolog 1, MLH1 , is due to hypermethylation of its promoter. The A allele of variant rs1800734 in the 5 ′ UTR of MLH1 modifies the binding of TFAP4 activating the BRAF/MAFG pathway, which increases DNMT3B-mediated methylation of the MLH1 promoter [ 98 ]. Another example of active genetic-methylation interplay is a mechanism suggested to underlie a type 2 diabetes (T2D) susceptibility locus [ 99 ]. The T allele of rs11257655 in the CAMK1D gene decreases DNA methylation in CAMK1D promoter as a meQTL, increases CAMK1D expression as an eQTL, and increases T2D risk as T2D GWAS signal. The authors propose that in the presence of the T allele at rs11257655, a protein complex formed by FOXA1/FOXA2 and other TFs binds to an enhancer of CAMK1D , which leads to demethylation of cg03575602 in the CAMK1D promoter and in turn upregulates its expression.
Trans -meQTL mechanisms
Many mechanisms have been hypothesized to underlie trans -acting meQTLs effects, but to date, very few clear examples have been uncovered. The simplest hypothesis is that SNPs that act as eQTLs of global methylation regulators, or their associated elements, have downstream effects as meQTLs at multiple CpG sites genome-wide (Fig. 3 a). For example, Lemire et al. [ 55 ] documented the case of SUMO-specific protease 7 (SENP7), which interacts with epigenetic repression proteins. Intronic variants located in SENP7 gene are cis -eQTLs, and high levels of the transcript decrease methylation at several trans -CpGs. Another case is variant rs12933229 associated with expression of RRN3P2 , a pseudogene that regulates DNA methylation through piwi-interacting RNAs (piRNAs).

Mechanisms underlying trans -meQLTs effects. a eQTL-mediated mechanism. If a SNP acts as an eQTL for a gene that regulates DNA methylation, the SNP can have an indirect effect on multiple CpG sites in trans . b Cis -meQTL-mediated mechanism. If a SNP is a cis -meQTL for nearby CpG sites, which in turn impact the expression of genes involved in epigenetic regulatory processes, the SNP can ultimately alter DNA methylation levels at CpG sites in trans . c 3D organization mechanism. In the 3D genome, distal sites can move in close proximity, whereby a SNP can affect a DNA methylation levels at CpG sites in trans , acting either through cis -meQTL mechanisms, or by disrupting the formation of structural loops. d SNPs in the coding regions of methyl-specific binding proteins (such as MeCP2) can alter their specificity and function, and therefore passively or actively (by recruiting DNMTs or TETs) modify DNA methylation of their binding sites
Other findings suggest that distal effects may be mediated, in total or in part, by cis -meQTL-associated CpGs (Fig. 3 b). For instance, one-third of the 585 trans -meQTL-CpG pairs identified by Shi et al. [ 56 ] in lung tissue showed weaker associations after conditional analyses, conditioning on the cis -regulated CpGs by the same SNPs. In 166 trans -meQTL associations, the authors found a partial mediation of cis effects, with lower but still significant partial correlations compared to marginal correlations, and in 30 associations, they found a full mediation, with no significant correlations after conditioning for cis -meQTLs. Genes for GTPase or related enzymes involved in DNA methylation regulation, were over-represented for such cis -CpGs. Therefore, one potential mechanism underlying trans -meQTL effects is that a meQTL may act on nearby CpGs, which then impact the expression of genes that eventually may modify DNA methylation levels at distal sites.
Three-dimensional (3D) genome conformation changes would be an alternative track for the action of trans -meQTLs, since distal loci can be brought into physical proximity by 3D structures [ 100 ]. Hence, either SNPs in TFBSs acting as cis -meQTLs, or SNPs in sites that anchor cohesins and CTCF that integrate topologically associating domains (TADs) and loops, could have an impact on remote CpGs as they move in closer proximity in complex 3D DNA structures (Fig. 3 c) [ 26 ]. Furthermore, the 3D organization of the DNA includes inter-chromosomal contact, which would be the source of a fraction of meQTLs associations—as demonstrated by high-resolution Hi-C data that CpGs overlap with binding sites of architectural proteins (e.g., CTCF, RAD21, and SMC3) [ 51 ], and with a two-dimensional functional enrichment [ 50 ].
Other explanations involve sequence-specific binding proteins, similar to mechanisms for cis -meQTLs, but instead of the genetic variant being located in TFBS, here, the SNPs interfere with the coding or cis -regulatory regions of the TFs, and thus their subsequent expression, coupling, and function (Fig. 3 d). The results of Bonder et al. [ 51 ] point in this direction. The authors found that 13.1% of the trans -meQTLs that they detected also altered the expression of TFs, and those affecting multiple CpGs had consistent direction of effects, either increasing or decreasing methylation at most of CpGs. A representative example is rs3774937 in the intron of the TF NFKB1 , which is a trans -meQTL for 413 CpG sites genome-wide. In 380 CpG sites, the rs3774937 alternative allele was associated with lower methylation levels, and 147 of those CpG sites were in NF- κ B binding sites.
The same mechanism could also apply to the activity of proteins other than sequence-specific binding proteins, although this theory remains mostly unexplored so far. For example, it is well known that DNA binding of some proteins is methylation-dependent through a methyl-CpG-binding domain (MBD), such as for the MeCP2 (methyl-CpG-binding protein 2). MeCP2 regulates DNMT3A allosterically, acting as a repressor or an activator of the methylation process [ 101 ]. However, some mutations in the MeCP2 gene decrease selectivity of the MeCP2 binding [ 102 ], and consequently, could lead to untargeted methylation at several distal sites. This idea can also be extended to proteins without MBD, as emerging evidence suggests [ 103 ]. This may also complement the sequence-specific binding sites theory and thus explain more trans -meQTLs.
MeQTLs and mechanisms underlying human disease
Many research efforts have linked meQTLs to genetic variation underlying human complex traits. MeQTLs are significantly enriched for GWAS signals, with evidence for shared genetic effects [ 50 ]. Multiple studies have explored the directionality of shared genetic associations, applying causal inference approaches typically exploring the potential role of DNA methylation as a mediator of genetic effects on phenotypes [ 38 , 50 , 104 ]. However, despite a substantial sharing of genetic effects, the findings reveal a more complex genetic architecture including putative evidence for both mediation effects of DNA methylation on phenotypes, as well as effects of complex traits on methylation (see the “ MeQTLs and GWAS ” section).
Nevertheless, the discovery of meQTLs has contributed to the advancement of our understanding of the molecular pathways underlying certain human phenotypic traits and diseases, which may eventually help towards the development of therapeutic targets. Examples include a thorough investigation of previously identified genetic signals for Alzheimer’s disease involving the promoter region of gene PM20D1 and meQTLs (rs708727–rs960603 haplotype) [ 105 ]. With a series of in silico, in vitro, and in vivo experiments, Sanchez-Mut et al. determined that meQTLs interact with the promoter of PM20D1 through haplotype-dependent 3D chromatin conformations via CTCF, changing DNA methylation levels, altering gene expression, and ultimately protecting or aggravating neurodegeneration. In another study focused on characterizing osteoarthritis risk variants in cartilage samples, Rice et al. [ 106 ] found four meQTLs for 17 CpGs. In vitro studies of the prioritized locus suggest potential DNA methylation and gene expression mechanisms altering the function of the PLEC and GRINA genes, which have not been previously described in context of osteoarthritis. Similarly, meQTLs have helped to elucidate biological pathways underlying other diseases such as Parkinson’s disease [ 107 ], multiple sclerosis [ 108 ], colorectal cancer [ 98 ], and T2D [ 99 ] (see the “ Cis -meQTL mechanisms ” section), along with complex phenotypes such as platelet function [ 109 ], fatty acid levels [ 110 ], and others.
Challenges and future directions
Methodological and statistical caveats, dna methylation profiling.
The vast majority of meQTL studies to date explore DNA methylation levels profiled by Illumina DNA methylation arrays, which are relatively low-cost and highly standardized. However, array-based DNA methylation profiles can be subject to bias introduced by errors from cross-hybridization events, as well as batch and positional effects. For example, positional effects have been reported to impact a larger proportion of 450K probes, compared to 27K probes [ 111 ]. In addition, both the 450K and EPIC arrays contain two different types of probes with different dynamic ranges [ 112 ]. Several methods have been developed to minimize bias introduced by these potential array effects [ 113 – 116 ], as well as comparisons across methods, which provide useful frameworks for the design of quality control and normalization of Illumina-based DNA methylation profiles [ 117 – 119 ]. Further work has also focused on guidelines for exclusion criteria of low-performing probes [ 120 – 122 ], or has explicitly flagged unreliable probes due to cross-reactive events or underlying genetic variation [ 123 ].
As previously discussed, genome coverage is a key consideration in DNA methylation profiling technologies, and here the ultimate aim is to characterize meQTLs across the entire methylome. With most studies based on array DNA methylation profiles, the EPIC array provides a reasonable cost-coverage balance with increased coverage of regulatory elements compared to the 450K. Despite the improvement in coverage by the EPIC array, regulatory regions included on the EPIC only comprise 27% of cis and 7% of trans regions characterised by ENCODE [ 123 ]. This, combined with the limited methylome coverage, should also be considered when generalizing meQTL findings to whole genome.
On the other hand, WGBS allows for comprehensive profiling of the methylome, but the high costs are still restrictive and prevent its broad application in meQTL studies. Also, some genomic regions and difficult to sequence and library preparation protocols are technically complex and may be subject to bias from multiple sources, such as bisulfite conversion, PCR amplification, DNA modifications, and degradation [ 124 , 125 ]. An important parameter to define in a WGBS experiment is the sequencing depth. The recommended depth coverage based on data from the NIH Roadmap Epigenomics Project [ 126 ] and the International Human Epigenome Consortium (IHEC) [ 127 ] is 30×. In order to optimize costs while maintaining acceptable rates of specificity and sensitivity, Ziller et al. [ 128 ] proposed a minimum coverage per sample of 5–15 × for the discovery of differentially methylated regions (DMRs). Nonetheless, coverage of 100× would be required to have similar precision to that in Illumina arrays [ 124 ]. In light of these estimates, WGBS in large-scale samples currently still poses significant challenges, but represents a promising method for future meQTL analyses, especially for studying regions of the genome underrepresented in microarrays.
Statistical models
The choice of statistical model for meQTL analysis is important. Most meQTL studies apply linear regression, but at many CpG sites the distribution of DNA methylation values does not meet its assumptions, which may in turn increase the error rate (both type I and II). Recently, Mansell et al. [ 129 ] quantified the extent of bias in epigenome-wide association studies (EWAS) using the EPIC array due to non-linearity between variables, non-normal distribution of residuals (skewness and kurtosis), and heteroskedasticity. The authors concluded that even CpG sites with extreme deviation to linear regression assumptions do not result in major bias. By extension, this observation could also apply to meQTL studies. Interestingly, the same study did not find better performance when using M -values instead of β -values in DNA methylation analysis. Ultimately, a higher selectivity of the CpG sites to test—such as filtering out probes with low β -values variability—would leverage the statistical confidence of the models and maximize reproducibility of results, as recommended by Logue et al. [ 122 ].
Multiple-testing correction
One major consideration in meQTL analyses is the multiple-testing correction, given the large number of tests in comparing millions of genetic variants against typically at least hundreds of thousands of CpG sites. On the one hand, the multiple testing correction must be computationally efficient, and on the other hand, the aim is to maximize statistical power to detect low or modest effects.
A considerable amount of studies apply permutation-based multiple testing thresholds to quantify an empirical false discovery rate (FDR) [ 130 ]. Typically, this approach consists of randomizing the genotypes to generate an approximate null distribution of p -values obtained in a large number of association tests between the permuted genotypes and CpG sites. The FDR is the ratio of associations in the permuted data to those observed at a specific nominal significance threshold [ 131 ]. Because permutations are computationally demanding, methods as FastQTL and QTLtools [ 132 , 133 ] have proposed variations of the original technique, such as drawing a few thousand permutations and modeling the resulting p -values with a beta distribution to approximate the null distribution. This approach has been adopted by some meQTLs analyses [ 83 , 93 ]. Moreover, some analyses have reduced the number of permutations, for example to a hundred [ 60 ] or even ten [ 51 , 82 ], a decision supported by eQTL results about the stability of the FDR value with as few as five permutations [ 134 , 135 ]. For example, cis -meQTL analysis by van Dongen et al. [ 39 ] in buccal cells of MZ twins applied the aforementioned method with ten permutations, conserving relatedness between twins by permuting twin pairs samples rather than individuals.
Other studies have applied the conservative Bonferroni multiple testing correction to control the family-wise error rate (FWER), adjusting for the total number of SNPs-CpGs pairs tested, resulting in stringent multiple testing significant thresholds (e.g., p <1×10 −10 ) [ 35 , 37 , 136 ]. However, the Bonferroni multiple testing correction does not take into account linkage disequilibrium (LD) between genetic variants or patterns of co-methylation, that is, the correlation in DNA methylation levels at nearby CpG sites [ 137 ]. To tackle LD, McRae et al. [ 53 ] and Hannon et al. [ 38 ] employed a Bonferroni threshold based on the GWAS canonical value 5×10 −8 —which accounts for LD blocks—and divided this threshold by the number of tested probes, while Smith et al. [ 86 ] adopted a Holm-Bonferroni method—or a step-down Bonferroni—which increases the power. To take into account co-methylation, it has been proposed that for the Illumina EPIC array in whole blood, an appropriate choice of the number of independent probes to control the FWER would be 530,639 (66% of total sites) [ 129 ].
MeQTL analysis strategies that use the Bayesian framework [ 138 ] or a multivariate normal distribution [ 139 ] have also been applied to other molecular QTL studies, and appear promising to explore in future meQTLs analyses.
Detection of common and rare variant meQTLs
Since publication of the first genome-wide meQTL studies, sample sizes have increased dramatically and with them power to detect small effects of common genetic variants on methylation. However, the detection of rare genetic variants is still a major challenge in meQTL studies. Almost all genome-wide meQTL studies discard SNPs with a minor allele frequency (MAF) less than 0.05 or 0.01, while the high penetrance of rare variants in certain complex traits highlights their biological importance [ 140 ].
The most widely implemented approaches for assessing effects of rare genetic variants on human complex traits are collapsing methods. The premise is that all the variants within the boundaries of a functionally meaningful locus would induce the same phenotypic change [ 140 ]. To our knowledge, only the study by Richardson et al. [ 141 ] has so far examined rare variants in meQTL analysis in blood samples. The authors collapsed variants with MAF ≤0.05 around CGIs (alone and with flanking shores/shelves) and carried out the Sequence Kernes Association Test (SKAT) testing for genetic influences on CpG sites from the 450K array. The results identified 94 unique cis -acting and one trans -acting regions, which were not previously linked to methylation. This novel approach can be leveraged in future meQTL analyses by the definition of other functional units for collapsing regions, testing previously identified meQTL regions after conditional analysis, and application to data from the EPIC array or WGBS.
Gene–environment interactions
Environmental exposures can leave a clear signature on DNA methylation patterns, as observed for smoking [ 18 , 142 , 143 ] and alcohol consumption [ 19 ]. Some environmental exposure or lifestyle factors and behaviors also have a genetic component that explains a proportion of their variance [ 144 , 145 ]. Therefore, to explore the interplay between genetic variation, environmental exposures, and epigenetic changes, some studies have considered gene–environment (G ×E) interaction terms in meQTL analyses.
To date and to our knowledge, no genome-wide G ×E analysis of DNA methylation has yet been published. However, G ×E analyses at candidate genomic regions have been described in several studies. For example, Teh et al. [ 146 ] studied the interaction of genetic effects and in utero environment in 237 umbilical cord samples in Asian neonates. Firstly, the authors identified 1423 variably methylated regions (VMRs) across individuals, based on the median absolute deviation of the DNA methylation levels in each CpG site. Then, to explore triggers of DNA methylation changes at each VMR, the authors assessed DNA methylation effects as a function of (1) genotype alone, (2) intra-uterine environment alone, or (3) the G ×E interaction. The intra-uterine environment was quantified through 19 parameters, including maternal smoking, maternal depression, and concentrations of compounds in maternal serum. Interaction models of genotype with different in utero environments had better performance at 75% of the VMRs compared to main-effects models, and therefore better explained the variability in DNA methylation. In two other studies [ 60 , 147 ] of monocyte samples from European and African populations, the authors suggested that some of the meQTLs may in fact be occurrences of G ×G and G ×E interactions (see the “ MeQTLs are differentially distributed across the genome ” section). The cis analysis uncovered 69,702 CpGs with meQTLs, and of these, 4.1% displayed different effects across the two populations, which may reflect G ×G or G ×E interactions.
Several studies have explored G ×E effects involving smoking status and genetic variants at candidate loci in the context of meQTLs and complex disease. Meng et al. [ 148 ] provide an example of a candidate G ×E effects linked to rheumatoid arthritis, involving genetic variants in the MHC and smoking status. They observed an effect of rs6933349 on cg21325723 (located in the body of the TSBP1 gene), only in current smokers. Further examples include the study of Klengel et al. [ 149 ] who investigated a G ×E interaction in FKBP5 , a gene that regulates the glucocorticoid receptor—a major component of the stress hormone system. The transcriptional activation of FKBP5 as a response to childhood abuse depends on genetic variants (rs1360780) that alter the 3D conformations of the locus; the expression of the gene is mediated by the demethylation in intron 7, a change that is long-term stable and has implications in stress disorders. A similar pathway potentially underlies methylation at SLC6A4 (serotonin transporter gene) [ 150 ].
The implementation of G ×E meQTL analyses entails challenges, such as substantially larger multiple testing burden and limited power [ 151 ]; however, it represents a promising niche to explore that may account for a fraction of missing heritability in DNA methylation.
MeQTL impacts on DNA methylation variance
Conventional statistical methods applied to QTL analysis aim to identify significant deviations of the trait mean between the subjects in different genotype groups, typically with the assumption of equal variance across groups (i.e., homoskedasticity). Recently, new perspectives in QTL studies have explored QTLs that influence the phenotypic variability across genotype groups, or variance QTLs (vQTLs/varQTLs). VarQTL may capture interaction effects, such as epistatic (G ×G) or G ×E [ 152 ]. Methods for detecting varQTLs include parametric and non-parametric tests, including Bayesian and family-based approaches [ 152 – 154 ]. One of the most comprehensive studies on varQTLs was carried out recently by Wang et al. [ 155 ], with genotype data from 348,501 participants from the UK Biobank and across 13 quantitative traits—including obesity-related, height, and lung function measures. The results show a total of 75 varQTLs for nine traits (54 varQTLs related to obesity) located in 41 nearly independent loci. Moreover, the authors found two varQTLs with possible non-additive effects on the variance, 66 varQTLs that also have an effect on the mean of the trait with the same direction, and 16 varQTLs that are explained by G ×E interaction models.
So far, only few studies have explored varQTLs in the context of molecular datasets, such as DNA methylation or gene expression profiles. For example, Brown et al. [ 156 ] examined variability of 13,660 genes in 765 LCL samples from the TwinsUK cohort, identifying 508 var-eQTLs in cis , of which 36% were also eQTLs. They then searched for variants interacting with each of the var-eQTLs within the same cis window in order to identify epistatic interactions, and found 256 G ×G signals, of which 57 replicated in another cohort. They also suggested that 70% of var-eQTLs may be the result of G ×E interactions based on analyses focusing on gene expression differences in MZ twins. In a methylome analysis with the 450K array in 729 peripheral blood leukocytes samples from individuals of Swedish descent, Ek et al. [ 157 ] estimated a total of 374,252 CpG-var-meQTL pairs, or 7195 unique CpGs with at least one var- cis -meQTLs. At almost all of these CpGs, there was also evidence of cis -meQTL effects, and after adjusting methylation levels for cis -meQTLs, the authors no longer found variance heterogeneity at the majority of CpGs. As a result, they conclude that a considerable proportion of varQTLs (92%) may be statistical artifacts attributed to SNPs in LD, rather than real biological interactions, and that var-meQTLs are unlikely to explain missing heritability.
Future studies are needed to replicate these var-meQTLs results, explore mechanisms driving these effects, and potentially identify novel signals.
Integrating meQTL results in association studies
Meqtls and ewas.
EWASs aim to systematically associate variation in DNA methylation levels across the genome with variation in phenotypes or environmental exposures. However, significant associations between DNA methylation levels and phenotypes may arise due to confounding effects of meQTLs, and most EWASs do not take meQTLs into account. Adjustment of DNA methylation values for meQTL effects prior to EWAS has been proposed to tackle this issue [ 158 , 159 ]. Chen et al. [ 78 ] applied this approach in EWASs of gene expression levels genome-wide, or in expression quantitative trait methylation (eQTM) analyses. The authors quantified the contribution of DNA methylation to gene expression variance through a variance decomposition model and found that DNA methylation explained a lower proportion of the variance in models adjusted for underlying genetic effects, compared to unadjusted models. Subsequently, they performed EWASs with two models—either not correcting for or correcting for cis -genetic effects. Over half of the genes associated with epigenetic marks in the uncorrected model did not reach significance in the corrected model. Although meQTLs effects were not directly assessed in this study, these findings may extend to meQTLs. In another study, Krause et al. [ 160 ] aimed to validate two candidate CpGs associated with T2D, but found a significant association between BMI and blood methylation only after correcting for genotype at rs9982016, which was a cis -meQTL at one of the candidate CpGs.
Another relevant application of integrating meQTLs in EWAS is to gain insight into the putative causal direction of association between DNA methylation signals and the associated phenotypes by using Mendelian randomization (MR). MR evaluates the likelihood that a phenotype is the consequence of an exposure, which in turn is the result of genetic variation (or the instrumental variable) [ 161 ]. In context of epigenetic analyses, meQTLs are instrumental variables, DNA methylation levels are exposures, and diseases or phenotypes are the outcomes [ 104 ]. Multiple studies have applied MR using meQTLs in EWAS across a range of phenotypes [ 162 – 166 ]. For example, in an EWAS of BMI in 3743 blood 450K methylomes from older adults and with replication, Mendelson et al. [ 167 ] identified 83 DMSs and their associated meQTLs. Follow on MR identified two CpGs (cg11024682 in SREBF1 , and cg07730360, unannotated) with nominally significant putative causal effects of DNA methylation on BMI. In contrast, they identified 16 CpG where DNA methylation levels are likely mediated by BMI after a reverse MR model. Using a similar approach, Dekkers et al. [ 168 ] analyzed if exposure to elevated blood lipids affected DNA methylation levels in immune cells, in 3296 450K methylomes from six Dutch biobanks. The authors identified 21 DMSs for triglycerides (TG) levels, three for low-density lipoprotein cholesterol (LDL-C) and four for high-density lipoprotein cholesterol (HDL-C). Follow on MR analysis identified putative causal effects of lipid levels on 13 DMSs. To exclude pleiotropy (SNPs acting as QTLs for multiple lipid levels, or as cis -meQTLs in DMSs) and reverse causation ( cis -meQTLs affecting DMSs, and DMSs affecting lipid levels), the authors conducted secondary MR analysis. The results confirmed that TG likely induced differential methylation at three CpGs, LDL-C at one, and either TG or HDL-C at two. Mendelian randomization has also been applied when integrating meQTL results and GWAS (see the “ MeQTLs and GWAS ” section).
In addition, the combination of EWAS and meQTL signals can be used to explore G ×E interactions. For instance, Tsaprouni et al. [ 169 ] found that almost half of smoking-associated loci have meQTLs. Subsequent analyses fitting G ×E interaction effects identified a CpG (cg03329539 located in chromosome 2) where methylation response to cigarette smoking was modulated by rs62192178 genotype.
MeQTLs and GWAS
Although thousands of GWAS results have been published to date, the identification of causal variants and their functional interpretation remains mostly outstanding. Furthermore, GWASs also face the “missing heritability” problem and epigenetic signals (potentially, through meQTLs) might explain a proportion of the phenotype missing heritability [ 27 , 170 ]. Therefore, integrating meQTL findings as a post-GWAS analysis can help to address some of these challenges.
One approach for this integration is to use meQTLs findings to prioritize GWAS signals for follow on analysis, for example, as applied in a study of autism spectrum disorders in 1263 infants by Hannon et al. [ 171 ]. The authors estimated that 91 SNPs associated with the disease were also meQTLs, based on a Bayesian co-localization analysis. Their results highlight specific variants to target in subsequent studies since they may have a functional role in autism pathophysiology. Morrow et al. [ 82 ] implemented a similar Bayesian framework to identify meQTLs that are also chronic obstructive pulmonary disease (COPD) GWAS signals (see the “ MeQTLs in non-blood-based tissues and cells ” section). Their findings identified 20 SNPs with suggestive evidence of co-localization, highlighting novel regions of interest in addition to previously identified COPD signals, such as KCNK3 and EEFSEC .
MR analyses have also been adopted to integrate meQTL and GWAS results. Richardson et al. [ 172 ] assessed putative causal effects at 30,328 CpGs in 139 complex traits based on previously published cis -meQTL and GWAS results. The authors assessed the fit of several models spanning: (1) a forward MR model where the DNA methylation level impacts the phenotype; (2) a joint likelihood mapping, to exclude genetic variants in LD independently influencing DNA methylation and phenotype; and (3) a reverse MR model to exclude cases where DNA methylation is the outcome. A final set of 346 CpG sites were identified as potentially causal across 46 traits, ultimately highlighting specific biological pathways and suggesting potential drug targets. Similar analyses have also been undertaken within specific phenotype domains by multiple other studies, including Huan et al. [ 35 ], Bonder et al. [ 51 ] and Chen et al. [ 78 ]. In the largest analysis so far, Min et al. [ 50 ] found a significant substantial enrichment of meQTLs with the GWAS signals in 13 of 37 phenotypes GWAS datasets assessed, especially for SNPs acting as both cis and trans -meQTLs. However, after multiple causal inference analyses, the authors observed that only for a minority of cases DNA methylation exhibited mediating effects of GWAS signals in complex traits, and vice versa. These directionality results have several interpretations, including the possibility that other molecular mechanisms may explain a proportion of the observed shared genetic signals.
Shared QTL effects on multiple regulatory genomic processes
Regulatory genomic changes capture multiple molecular processes across different layers of epigenetic data. Comparison of meQTLs with QTLs for different biological profiles is a promising route to infer regulatory potential. In spite of the considerable amount of studies that jointly consider DNA methylation and gene expression data, relatively few studies have explicitly compared eQTLs and meQTLs genome-wide. Such comparisons have been based on either summary statistics of published studies [ 92 , 173 ] or de novo associations [ 49 , 51 , 59 , 67 , 78 , 83 , 93 ]. Overlapping results can be used to identify pleiotropic effects for DNA methylation and expression and explore directionality of these effects, such as SNP →methylation →expression (active) or SNP →expression →methylation (passive). For example, Gutierrez-Arcelus et al. [ 59 ] inferred that DNA methylation can have both active and passive roles in gene expression regulation across fibroblasts, T cells and lymphoblastoid cells from the umbilical cords of 204 babies. Furthermore, comparison of meQTLs with other epigenetic data QTLs may also give further insights into regulatory epigenetic processes. Banovich et al. [ 67 ] compared meQTLs with QTLs for histone modifications, PolII occupancy and DNAse I hypersensitivity, and based on the extent of overlap observed they hypothesized that coordinated regulatory changes may be explained by modified TF binding affinities. Chen et al. [ 78 ] explored similar questions in three different immune cell types ( n =525), where 43.3% of the genetic variants identified as eQTLs were either found to have a coordinated effect as meQTLs or to be in high LD with a meQTL. However, the effect sizes were weakly negatively correlated, which the authors interpreted as a partial uncoupling between methylation and expression. The study also included analysis of histone modification QTLs (hQTLs for H3K4me1 and H3K27ac), where again 43.3% of eQTLs and hQTLs overlapped with strong positive correlation in effect sizes, suggesting an active role for histone modifications on expression.
As additional data are being generated on multiple epigenetic and expression layers of data, future analyses will have greater power to explore the regulatory nature of meQTLs. However, co-localization results should be interpreted with caution, as the intersection of QTLs does not imply a causal relationship or direct association due to LD or statistical artifacts. Additionally, if summary statistics are obtained from databases with different reference populations, the significant signals may not be comparable [ 25 ].
In conclusion, the identification of methylation quantitative trait loci genome-wide has significantly increased our knowledge of the factors driving DNA methylation variation in humans, and holds value for integrating genomics and epigenomics in the context of disease.
Acknowledgements
SV acknowledges the financial support of the Mexican National Council of Science and Technology (CONACYT).
Review history
The review history is available as additional file 1 .
Authors’ contributions
Both authors wrote and approved the final version of the manuscript.
The work was supported by the ESRC (ES/N000404/1 to JTB), JPI-HDHL through BBSRC (BB/S020845/1 to JTB), and CONACYT doctoral fellowship (2019-000021-01EXTF-00323 to SV).
Declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sergio Villicaña, Email: [email protected] .
Jordana T. Bell, Email: [email protected] .
REVIEW article
This article is part of the research topic.
Pathogen-Induced Immunosenescence: Where do Vaccines Stand?
TB and HIV Induced Immunosenescence: Where do vaccines play a role? Provisionally Accepted

- 1 Western University of Health Sciences, United States
- 2 Chamberlain University, United States
The final, formatted version of the article will be published soon.
This paper tackles the complex interplay between Human Immunodeficiency virus (HIV-1) and Mycobacterium tuberculosis (M. tuberculosis) infections, particularly their contribution to immunosenescence, the age-related decline in immune function. Using the current literature, we discuss the immunological mechanisms behind TB and HIV-induced immunosenescence and critically evaluate the BCG (Bacillus Calmette-Guérin) vaccine's role. Both HIV-1 and M. tuberculosis demonstrably accelerate immunosenescence: M. tuberculosis through DNA modification and heightened inflammation, and HIV-1 through chronic immune activation and T cell production compromise. HIV-1 and M. tuberculosis co-infection further hastens immunosenescence by affecting T cell differentiation, underscoring the need for prevention and treatment. Furthermore, the use of the BCG tuberculosis vaccine is contraindicated in patients who are HIV positive and there is a lack of investigation regarding the use of this vaccine in patients who develop HIV co-infection with possible immunosenescence. As HIV does not currently have a vaccine, we focus our review more so on the BCG vaccine response as a result of immunosenescence. We found that there are overall limitations with the BCG vaccine, one of which is that it cannot necessarily prevent reoccurance of infection due to effects of immunosenescence or protect the elderly due to this reason. Overall, there is conflicting evidence to show the vaccine's usage due to factors involving its production and administration. Further research into developing a vaccine for HIV and improving the BCG vaccine is warranted to expand scientific understanding for public health and beyond.
Keywords: immunosenescence, M. tuberculosis, Vaccine3, BCG, HIV
Received: 14 Feb 2024; Accepted: 13 May 2024.
Copyright: © 2024 Singh, Patel, Seo, Ahn, Shen, Nakka, Kishore and Venketaraman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
* Correspondence: Mx. Vishwanath Venketaraman, Western University of Health Sciences, Pomona, 91766-1854, California, United States
People also looked at
IMAGES
VIDEO
COMMENTS
DNA (deoxyribonucleic acid) is the nucleic acid polymer that forms the genetic code for a cell or virus. Most DNA molecules consist of two polymers (double-stranded) of four nucleotides that each ...
DNA Research is an internationally peer-reviewed journal which aims at publishing papers of highest quality in broad aspects of DNA and genome-related research ... Accepting high quality papers on broad aspects of DNA and genome-related research. Submit. Related Titles.
Genomic research has evolved from seeking to understand the fundamentals of the human genetic code to examining the ways in which this code varies among people, and then applying this knowledge to ...
This comprehensive review paper consolidates research efforts, focusing on DNA repair mechanisms, computational research methods, and associated databases. Our work is a valuable resource for scientists and researchers engaged in computational DNA research, offering the latest insights into DNA-related proteins, diseases, and cutting-edge ...
The information encoded by DNA is both digital - the precise base specifying, for example, amino acid sequences - and analogue. The latter determines the sequence-dependent physicochemical properties of DNA, for example, its stiffness and susceptibility to strand separation. Most importantly, DNA chirality enables the formation of supercoiling ...
Introduction. DNA synthesis occurs during the S phase of the cell cycle and is ensured by the replisome, a molecular machine made of a large number of proteins acting in a coordinated manner to synthesize DNA at many genomic locations, the replication origins 1.Replication origin activation in space and time (or replication program) is set by a sequence of events, starting already at the end ...
Effective DNA synthesis is therefore vital to close the gap between the ability to read and write DNA. Fig. 1: The state of the art in DNA synthesis. a, Productivity of DNA reading and DNA writing ...
Genetic functions of DNA can be understood in two ways: the nitrogen base sequence, constituting the archive of information encoding the sequences of proteins and RNA, and the double helix ...
Polymerase chain reaction (PCR) is a robust technique to selectively amplify a specific segment of DNA in vitro . [ 1] PCR is performed on thermocycler and it involves three main steps: (1) denaturation of dsDNA template at 92-95°C, (2) annealing of primers at 50-70°C, and (3) extension of dsDNA molecules at approx. 72°C.
Synthetic biology is a research area that is emerging rapidly and impacting extensively on biology, nanofabrication, and medicine. ... A survey paper on DNA-based data storage. 2020 International Conference on Emerging Trends in Information Technology and Engineering (Ic-ETITE), IEEE (2020), pp. 1-4.
DNA extraction, playing an irreplaceable role in molecular biology as it is an essential step prior to various downstream biological analyses. Thus, the accuracy and reliability of downstream research outcomes depend largely on upstream DNA extraction methodology. However, with the advancement of do …
1. Introduction. The potential benefits of DNA profiling are clear. When two DNA profiles do not match, it suggests that the DNA samples were derived from different individuals; it provides exclusionary evidence [1].Post-conviction DNA comparison has helped to exonerate many from wrongful convictions [2].Forensic DNA analysis, based on the analysis of a single or simple mixed DNA sample, has ...
Biotechnology which is synonymous with genetic engineering or recombinant DNA (rDNA) is an industrial process that uses the scientific research on DNA for practical applications. rDNA is a form of ...
Invert the tube containing the DNA pellet on tissue paper to complete draining off the supernatant. xi. Wash DNA pellet with 500 μl of 70% ethanol and invert once (to dissolve residual salts and to increase purity of the DNA). xii. ... (MJ Research PTC-100 thermocycler) programmed to perform an initial denaturation step of 98 °C for 2 min ...
The DNA polymerases that appear most able to tolerate modifications include KOD Dash, ... (GM060005 and GM097489) and the Defense Advanced Research Projects Agency Folded Non-Natural Polymers with Biological Function Fold F(x) Program (Award No. N66001-14-2-4052). ... Papers of particular interest, published within the period of review, have ...
Current research efforts to quantify DNA transfer and isolate the factors influencing its variability are summarized here. The third question concerns the biological or cellular origins of the DNA recovered from touched objects and remains largely unanswered [10]. The fundamental question of precisely where "touch DNA" is coming from has ...
Current research in forensic DNA and genetic analysis have focused on the reconstruction of the physical appearance of individuals—DNA phenotyping (Kayser, ... In this focus paper, we reviewed the major reasons that may limit the effectiveness of forensic DNA analysis, including the scope of its application, significance or usefulness ...
MLA. APA. Chicago. University of California - Santa Cruz. "Cellular activity hints that recycling is in our DNA." ScienceDaily. ScienceDaily, 10 May 2024. <www.sciencedaily.com / releases / 2024 ...
AlphaFold 3 can predict how DNA, RNA, and other molecules interact, further cementing its leading role in drug discovery and research. Who will benefit? Google DeepMind has released an improved ...
DOI: 10.1021/jacs.4c03413. A new technique in building DNA structures at a microscopic level has the potential to advance drug delivery and disease diagnosis, a study suggests. A team of ...
DISCOVERY OF THE DNA FINGERPRINT. Historically, identity testing in the forensic field started with the analysis of the ABO blood group system. Later, new markers for identity and paternity identification were based on variations of serum proteins and red blood cell enzymes; eventually the human leukocyte antigen system was used ().It was not until 20 years ago that Sir Alec Jeffreys ...
A new research paper was published in Aging, titled, "The association between neighborhood deprivation and DNA methylation in an autopsy cohort." Previous research has found that living in a ...
Move over, chatbots. This upgraded AI can model antibodies, DNA, and molecules from disease organisms. This next generation of AlphaFold, from Google Deepmind, is poised to significantly advance ...
AlphaFold 2 and 3. Debuted in late 2020, AlphaFold 2 solved a grand scientific challenge because it was able to accurately predict the structure of most proteins simply from their DNA sequence.
Unlike DNA sequence, genomic methylation patterns are not directly inherited during meiosis [], but are mostly reprogrammed in two waves during embryogenesis [13-15].Following this, DNA methylation modifications can be both stable and dynamic during mitosis events that accumulate over the life course [16, 17].These observations suggest that the environment may be a key driving force behind ...
This paper tackles the complex interplay between Human Immunodeficiency virus (HIV-1) and Mycobacterium tuberculosis (M. tuberculosis) infections, particularly their contribution to immunosenescence, the age-related decline in immune function. Using the current literature, we discuss the immunological mechanisms behind TB and HIV-induced immunosenescence and critically evaluate the BCG ...