PhD in Medicinal Chemistry
Application deadline for incoming class of 2024: December 15th, 2023 Prospective student visits (by invitation only): Late Feb 2024

The Department

We have 8 faculty running active research labs tackling health-related problems at the interface of chemistry and biology. Our department is internationally recognized for work on the mechanism and kinetics of xenobiotic metabolism by cytochrome P450s (CYPs) and other detoxification enzymes. CYPs metabolize most drugs used in the clinic, and their dysfunction is linked to harmful drug-drug interactions, inflammation, cancer, heart disease, and impaired neurodevelopment. Recently, our research has diversified into other areas of biochemistry, pharmaceutical chemistry, biophysics and chemical biology. Some faculty study therapeutic antibodies, peptides and other biologics, which are revolutionizing clinical practice. Other faculty study virology with the goal of developing better vaccines and treatments for diseases like HIV/AIDS and influenza, or neurodegeneration with the goal of improving the diagnosis and treatment of dementias like Alzheimer’s disease. Our faculty also develop new analytical techniques, such as mass spectrometry methods to characterize lipids, metabolites and glycoproteins more quickly and sensitively than ever before. Learn more about each lab’s research here .
The PhD Experience

Coursework requires students to become proficient in organic, medicinal and physical chemistry, pharmacology, biochemistry, and molecular biology. (See a typical Program of Study , and course descriptions in the course catalog .) The curriculum is adaptable to individual interests and needs, and most didactic coursework is completed in the first two years. For more information on the program please refer to our most recent Med Chem student handbook .
Professional development outside the laboratory and classroom is a major point of emphasis. Students build communication skills through regular presentations to their labs, and in departmental journal clubs and research seminars. Many students interested in biotech/pharma careers have benefited from our innovative industry mentorship and internship programs.
Financial Support
Incoming graduate students are generally supported by a research assistantship from the department for the first year of study, allowing students to dedicate their time to study and work in the lab. It currently covers tuition (excluding a $265 per quarter student fee) and an additional stipend of $3259 /month. In subsequent years, support is provided either by the department or by research or training grants. Outstanding applicants are considered for an ARCS Scholarship that provides an additional stipend of $7,500 for the first year and $5,000 for the next two years of graduate school. The research assistantship also provides health insurance at no charge for students; coverage is available for spouses and dependents for an additional fee. (You can find more information on the Graduate Appointee Insurance Program and other benefits through UW Human Resources .)
Career Opportunities
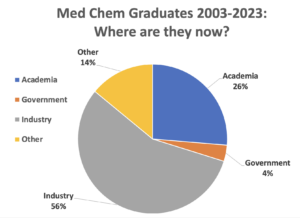
Our graduates also excel in academia. Recent graduates have done post-doctoral work at institutes such as Yale University, University of California San Francisco, University of California San Diego, Lawrence Berkeley National Lab, Queen Mary University of London, and Children’s Hospital Research Center. Subsequently, alums have gone on to tenure-track faculty positions at top-tier research universities, colleges of pharmacy, and liberal arts colleges.
More Application Info & FAQs
- Department of Chemistry >
- Graduate >
- Graduate Overview >
PhD in Medicinal Chemistry

Find your home in UB Chemistry! We're here to help you every step of the way.
- 3/5/24 Graduate Admissions
- 12/7/23 Financial Aid and Funding

Already enrolled in UB? Get details about advisement, forms and other resources for current students.
- 5/25/23 Info for Current Students

The PhD in Medicinal Chemistry provides a unique opportunity for students to develop a strong foundation in organic and medicinal chemistry and also to broaden their knowledge in areas such as drug discovery, biochemistry, molecular biology and pharmacology.
PhD Program Requirements
- Coursework Once admitted to the PhD in Chemistry program, students are required to complete six graduate-level lecture courses during the first two years of full-time study. Of these courses, three must be one-semester introductory core courses selected from the four traditional areas of chemistry (CHE 501 and MCH 501 are required for the Medicinal Chemistry PhD), while the other three elective courses are chosen in consultation with the student’s research advisor.
- Proficiency Students must also demonstrate proficiency in medicinal chemistry, as well as in three of four traditional areas of chemistry, during the first three semesters. Proficiency can be established by completing a core graduate course or by passing the ACS Placement Exam in the area. A 3.00 grade point average in lecture courses is required.
- Research Synopsis During the fifth semester (third year) of graduate study, PhD students are required to prepare a written research synopsis summarizing research progress to date and future research plans. An oral examination with the student’s PhD committee is used to evaluate the student’s research potential.
- Research Proposal Also during the fifth semester, the student is required to write and orally defend an independent research proposal. This proposal involves the identification of a problem from the chemical literature that is not directly related to the student’s thesis work and a proposed solution to that problem. There are no cumulative exams in the UB Department of Chemistry.
- Public Lecture During the fourth year of graduate study, PhD students present a public lecture on their research progress. This provides the PhD committee a chance to give the student feedback prior to finishing their written dissertation.
- Dissertation and Oral Defense The majority of a PhD student’s time is spent on creative research. At the conclusion of the research work, a dissertation must be written and orally defended before the PhD committee and the department at large.
Faculty Research Mentor
The Department of Chemistry views an advanced degree in chemistry or medicinal chemistry as primarily a research degree, so the choice of research director is an important decision for the first-year graduate student. To facilitate the selection of the research mentor, the members of the faculty engaged in research present a general overview of their research interests in a series of meetings with the new graduate students. This allows the students to become acquainted with the different research opportunities in the program in an informal setting.
Students are also encouraged to speak informally with as many faculty members as possible before making their decision. Assistance is available to those students having difficulty with this decision. However, it is to the student’s advantage to select a research advisor at the earliest possible date. Typically, graduate research is initiated during the second semester or during the first summer within the program.
PhD Student Timeline
Upon arrival, all new graduate students are required to take standardized tests produced by the American Chemical Society to assess their preparation for graduate study. Results of these tests are used by the Graduate Curriculum Committee to help students select their first-semester courses. A typical first-semester graduate student takes three core graduate-level courses and is also engaged in TA duties. Most of the required course work is finished by the end of the second or third semester in the program.
The following table provides a typical PhD graduate student timeline:
Email [email protected] or contact Prof. Timothy Cook , director of graduate studies, for more information on this program and the admissions process.
The PhD Program in Medicinal Chemistry educates and trains students in the design and synthesis of novel, biologically active compounds and in delineating their mechanisms of action using biochemical, biophysical, and pharmacological approaches. Research specializations are available in synthetic, biochemical/pharmacological, and biophysical aspects of medicinal chemistry. Doctoral research in these specializations will relate to faculty areas of research, which currently include substance use disorders and addiction; neuropathic pain; obesity and metabolic disorders; neuropsychiatric disorders (psychoses, ADHD, depression, anxiety, eating disorders); and neurodegenerative diseases.
In The News

Cannabis Will Transform Medicine—Once We Figure Out How to Get Rid of Its Side Effects

Drug Discovery Spurs Innnovation, Collaboration

Groundbreaking Cancer Gene Therapy
This specialization offered by the Center for Drug Discovery (CDD) trains students in the design and synthesis of novel biologically active compounds and in the study of their mechanisms of action using biochemical, biophysical, and pharmacological approaches. Concentrations are available in synthetic, biochemical/pharmacological, and biophysical medicinal chemistry. The CDD’s excellence in teaching has been recognized by the award of a training grant from the National Institute on Drug Abuse for predoctoral and postdoctoral training in development of medications. These will be targeted to treat drug abuse; addiction; and other indications such as neuropathic pain, obesity, neuropsychiatric disorders (psychoses, ADHD, depression, anxiety, eating disorders); and neurodegenerative disorders.
Where They Work
- Novartis Institutes for Biomedical Research
What They Do
- Healthcare Services
- Business Development
What They’re Skilled At
- High-Performance Liquid Chromatography
- Pharmaceutical Industry
- Cell Culture
- Biotechnology
Application Materials
Application.
- Application fee – US $50
- Three letters of recommendation
- Transcripts from all institutions attended
- Personal Statement
- Official GRE scores
- TOEFL score for applicants who do not hold a degree from a U.S. institution and whose native language is not English
- Please note all international applicants will need to provide a WES evaluation. Link to WES: https://www.wes.org/ https://www.wes.org/
Admissions deadline for Fall term: December 6
- Program Website
Request Information for PhD in Medicinal Chemistry and Drug Discovery
Academic Catalog 2023-2024
Medicinal chemistry and drug discovery, phd, journal club participation, colloquium attendance, internship requirements and regulations for department of pharmaceutical sciences, qualifying examination, doctoral candidacy status, doctoral dissertation committee, dissertation proposal defense, registration for dissertation, publications and presentations, phd dissertation preparation, pharmaceutical sciences colloquium, sopps professional code of conduct .
The PhD Program in Medicinal Chemistry and Drug Discovery educates and trains students in the design and synthesis of novel, biologically active compounds and in delineating their mechanisms of action using biochemical, biophysical, and pharmacological approaches. Research specializations are available in synthetic, biochemical/pharmacological, and biophysical aspects of medicinal chemistry. Doctoral research in these specializations will relate to faculty areas of research, which currently include substance use disorders and addiction; neuropathic pain; obesity and metabolic disorders; neuropsychiatric disorders (psychoses, ADHD, depression, anxiety, eating disorders); and neurodegenerative diseases.
The Department of Pharmaceutical Sciences sponsors weekly journal clubs, Pharmaceutical Science Seminar ( PHSC 6300 ) , at which students present and evaluate current scientific literature in their fields of study. Students must attend one of these journal clubs (Pharmaceutics & Drug Delivery Journal Club, Pharmacology Journal Club, or Medicinal Chemistry & Drug Discovery Journal Club), chosen in consultation with their advisors.
Attendance at one of these journal clubs is required each and every academic semester, as an integral part of the PhD curriculum, with the exception of the last year (year four) in the program. All PhD students must participate full-time in journal club for course credit, Pharmaceutical Science Seminar ( PHSC 6300 ) , for six semesters. Failure to attend journal club regularly may result in sanctions such as probation or dismissal from the PhD program. Any student who does not comply with these (or any other) conditions required in the PhD program faces potential dismissal.
All PhD students, regardless of program, are required to attend the weekly Pharmaceutical Science Colloquium series. Announcements of times and locations will be distributed weekly to students by email to their university email addresses. Attendance is recorded by sign-up sheet. One excused absence is permitted per semester. Failure to attend colloquia may result in sanctions such as probation or dismissal from the PhD program.
Internships provide an experiential component of the graduate curriculum that fosters professional development through work in the pharmaceutical and biotechnology industries.
After PhD candidates have completed their dissertation research and are working on their dissertations, they are able, with the express permission of their PhD advisor, to participate in an internship if they choose. They are never allowed to intern while they are serving as teaching assistants.
- Students are responsible for finding their own internship and must be honest and accurate representing their experiences on their resumés. Students are responsible for tracking this experience on their resumés as there will be no detailed record on students’ transcripts of these opportunities.
- In order to be eligible for internship, students must take Professional Development for Pharmaceutical Sciences ( PHSC 5305 ) a semester before internship.
- Students must not accept more than one position. They must honor the first offer accepted. Any student not adhering to this requirement will not be allowed to participate.
- International students must register for Pharmaceutical Science Internship ( PHSC 6401 ) and follow instructions to receive Curricular Practical Training authorization from the Office of Global Services every semester they work. This applies to part-time jobs and volunteer opportunities. International students cannot engage in full-time CPT authorization totaling more than 52 weeks. Doing so will eliminate the possibility of engaging in the postgraduation benefit of Post-Completion Optional Practical Training.
- In order to receive a grade for the course, students must write at least two learning goals within the first two weeks of the internship and a one- to two-page paper describing what they learned, mid- and end of semester. Supervisors for internships will reply to a questionnaire about students’ performance.
- Taking internship must not extend international students’ visas.
- There are no vacations on co-op/internships. Companies’ sick time policies may vary. Students should check with their employers. For all other matters, please see the University-wide Academic Policies and Procedures and/or Bouvé College of Health Sciences Academic Policies and Procedures .
The PhD qualifying examination is required for students in all four programs under the auspices of the Department of Pharmaceutical Sciences: pharmacology, medicinal chemistry and drug discovery, biomedical sciences, and pharmaceutics and drug delivery. Students from each of the four programs will take the exams within the same time frame (below), regardless of specialty-area program focus.
Doctoral students should have selected a dissertation advisor by the end of their first year in the program and are expected to have begun research and demonstrated initial proficiency in the laboratory before taking the PhD qualifying examination.
The PhD qualifying examination tests the candidates’ knowledge and skills in core courses and program content areas. The overall PhD qualifying examination consists of two written exams and one oral exam. The qualifying examination is taken as a course, Doctoral Training and Research ( PHSC 8940 ) , no later than during the fall semester of the student's second year, after having successfully completed all the core courses of their respective programs.
At least two departmental faculty will contribute questions for the written exams, and no one faculty member will write more than the equivalent of one entire exam. All students qualified to sit for the exams are expected to take them at the times announced.
The format for the written exams may vary (e.g., faculty may ask a series of comprehensive essay questions or provide research publications(s) from the biomedical literature and ask questions based upon the publications’ content). The first exam is given in the first week of fall semester, with the written portion of the second exam (i.e., the F31 written document) to be submitted to the student’s exam committee by end of October with the oral presentation to be completed by mid-November and graded by the providers of the question(s).
- For example, if the student is in the pharmaceutics and drug delivery PhD program, part 1 will be about pharmaceutics and drug delivery, and part 2 can focus either on pharmacology or medicinal chemistry and drug discovery.
- Written exam 2 requires that students write an NIH F31 grant proposal and have the proposal signed off as passing by their examination committee after an oral defense.
A score of at least 70% is required to pass the first written exam (two parts). Students must pass all written portions of the PhD qualifying examination prior to the oral defense of the F31 proposal. Students who fail one written exam will have one opportunity to retake and pass that examination. A student who fails the first exam twice will be required to withdraw from the PhD program.
During the oral exam, students defend their NIH F31 grant proposal before an examination committee of, minimally, four faculty members: the dissertation advisor, at least two other Department of Pharmaceutical Sciences faculty members, and at least one member from outside the department. This committee is convened only for the oral exam and does not need to be the same committee as the student's dissertation committee.
Members of the oral examination committee are selected by the student, after consultation with the dissertation advisor and/or the director of graduate studies. The oral exam is graded on a pass/fail basis. Students who fail the oral exam on the first attempt may retake the exam within a time period designated by the examination committee not to exceed two months from the first oral exam. Those who fail twice will be dismissed from the program.
Doctoral students who have completed satisfactorily and thereby earned the credits for all required core courses (including those for their specialized area) and who have passed the written and oral qualifying examinations shall be admitted to candidacy status for the PhD degree.
Doctoral students must complete a dissertation that embodies the results of extended research and makes an original contribution to their field. This work should give evidence of candidates’ abilities to conduct independent investigation and interpret the results of their research in a professional manner. The doctoral dissertation advisor serves as chairperson of the Doctoral Dissertation Committee, which consists of no fewer than five members. Selection of an advisor is by mutual consent of the student and a member of the faculty, with approval by the director of graduate studies in the Department of Pharmaceutical Sciences. At least two members of the Doctoral Dissertation Committee must be faculty members in the Department of Pharmaceutical Sciences. At least one member is to be selected from outside the department. Committee members are chosen for their expertise in students’ research areas.
Within a year after successful completion of the PhD qualifying examination, but no later than the beginning of the fall semester of the third year, students must prepare and defend a written proposal detailing their planned dissertation project. Failure to do so will be regarded as a failure to progress in the PhD program and will result in a warning from the director of graduate studies of the Department of Pharmaceutical Sciences.
Students who do not correct this deficiency within one semester will be placed on academic probation. Students on academic probation must complete the dissertation proposal defense and return to nonprobationary status within one semester or be dismissed from the PhD program.
The dissertation proposal should be no more than 50 double-spaced pages (12-point font minimum and one-half-inch margins on all sides). This page limit excludes references but includes figures, figure legends, and tables. Aside from these exceptions, the proposal should otherwise conform to the format and structure of an NIH grant proposal with four main sections: specific aims, background and significance, preliminary studies, and experimental design and methods. The Department of Pharmaceutical Sciences Dissertation Proposal document provides detailed instructions on the preparation of a dissertation proposal. Associated required forms may be found on the SOPPS Student Portal Canvas site.
The dissertation proposal must be defended orally before the student's dissertation committee and signed by all dissertation committee members in approval of the student's planned dissertation research. Upon dissertation approval, the copies of the signed proposal approval cover sheet must be submitted to the department’s director of graduate studies and to the Bouvé College of Health Sciences Graduate Office.
Biannual Review
Dissertation committees meet routinely at six-month intervals, but no less than once a year, to evaluate students’ research progress and to be presented with written and oral progress reports on the direction and status of the research. Progress reports should be written in a brief format, identical to that described for the formal dissertation (see instructions listed on the SOPPS Student Portal Canvas site). Unsatisfactory productivity provides the basis for a warning by the dissertation committee and/or the Graduate Committee. Two such warnings will result in a student’s dismissal from the program.
Advisor consent and completion of all coursework (with the exception of the colloquium course) must be documented before students register for the first dissertation course. Students must register for Dissertation Term 1 ( PHSC 9990 ) and Dissertation Term 2 ( PHSC 9991 ) . Students must register for Dissertation Continuation ( PHSC 9996 ) each semester thereafter until the dissertation has been successfully defended. The department strongly encourages PhD students to complete the program within five years after acceptance, i.e., by three years after establishing degree candidacy. According to university policy, no PhD students may remain in the program for more than seven years.
Prior to completion of PhD training, candidates must present their research either as a poster or podium presentation at a regional or national scientific conference. Also prior to completion, the student must have submitted (preferably, published) at least one manuscript in a peer-reviewed journal that reflects original findings and laboratory work from the candidate's dissertation research.
Detailed guidelines for the format and content of the written dissertation are given in Instructions for Preparation of the Dissertation found on the SOPPS Student Portal Canvas site. The completed dissertation document should be reviewed first by the dissertation advisor. Feedback from the advisor should be incorporated into the dissertation draft before its distribution to the dissertation committee. The completed dissertation should be delivered to all dissertation committee members no later than two weeks before the scheduled oral defense.
All PhD candidates nearing completion of their research are required to present their dissertation findings at the department’s Pharmaceutical Sciences Colloquium. These presentations should be scheduled at least six months before anticipated completion of the dissertation. In turn, the dissertation should be completed no later than one year after the colloquium presentation. Students must register for Pharmaceutical Science Colloquium ( PHSC 6810 ) during the semester that the colloquium presentation is to be given.
Oral Dissertation Defense
The oral dissertation defense takes place after students complete their PhD dissertation research and all other requirements for the PhD degree. The oral defense deals with the subject matter of the dissertation, significant developments in the field, and students’ background knowledge in their field of concentration.
The dissertation committee conducts the final defense. The committee may recommend that the student clarify, amplify, or rewrite portions of the dissertation before the final defense is scheduled. Once the committee concurs that that written dissertation document is acceptable, a date is chosen for the final oral examination.
At least two weeks prior to the defense, students should inform the director of graduate studies in the Department of Pharmaceutical Sciences of the date of defense, so that advance announcement may be distributed. The final defense is open to anyone who wishes to attend and typically lasts at least two hours. After presentation of the work by the student in a seminar format, and responses to audience and committee questions, the committee meets first with the student for any follow-up discussion and then in executive session to decide whether the student has defended the dissertation successfully.
The committee’s decision is then announced to the student. If the committee’s vote is favorable, the student incorporates committee suggestions and corrections, if applicable, and the dissertation is signed and passed on to the department’s director of graduate studies. Requests for a second defense are highly irregular but may be permitted in the event that the previous oral defense was judged by the committee to be highly promising but inadequate in one critical aspect.
The final dissertation must be written, defended, and approved at least two weeks before the university commencement deadline. Students must submit signed copies of their dissertations to the website designated by the university and must abide by any embargo sanctioned by the student’s principal dissertation advisor and/or dissertation committee. The students should apply for graduation before the final dissertation defense, on the assumption that the dissertation will be approved. If the dissertation committee decides that more time is required to complete the dissertation beyond the commencement date, then the application for graduation can be withdrawn and a new one submitted pending final dissertation approval.
All SOPPS students (BSPS, Preprofessional, MS, and PhD) are expected to adhere to the Code of Conduct .
Please visit Bouvé College of Health Sciences Program Learning Outcomes for the specific student learning outcomes for this program.
Complete all courses and requirements listed below unless otherwise indicated.
Qualifying examination Doctoral candidacy status Doctoral dissertation committee Dissertation proposal Biannual review Pharmaceutical Sciences Colloquium Oral dissertation defense
Core Requirements
A grade of C– or higher is required in each course.
Research and Dissertation
Program credit/gpa requirements.
32 total semester hours required Minimum 3.000 GPA required
Plan of Study
Scientific Writing: Thesis Proposal ( PHSC 7020 ) must be taken the summer before the qualifying exams.
Doctoral Proposal ( PHSC 9681 ) should be taken in summer of second year, but no later than fall of third year.
Pharmaceutical Science Colloquium ( PHSC 6810 ) must be taken six months before dissertation defense.
PHSC 5305 & PHSC 6213 is suggested to be taken in the fourth year, but can be taken at any point before graduation.
Plan of Study - Advanced Entry
Doctoral Proposal ( PHSC 9681 ) may be taken in spring of first year but must be taken before fall of second year.
Pharmaceutical Science Colloquium ( PHSC 6810 ) must be taken six months before dissertation defense.
Advanced entry into the Medicinal Chemistry and Drug Discovery PhD program requires a master's degree in pharmaceutical sciences or a related area and focuses on various advanced research courses and successful defense of the dissertation. An applicant's transcripts are required to be reviewed by the admissions committee to ensure they are eligible to be in the advanced entry program.
Annual review Qualifying examination Dissertation committee Dissertation proposal Dissertation defense
10 total semester hours required Minimum 3.000 GPA required
Print Options
Send Page to Printer
Print this page.
Download Page (PDF)
The PDF will include all information unique to this page.
2023-24 Undergraduate Day PDF
2023-24 CPS Undergraduate PDF
2023-24 Graduate/Law PDF
2023-24 Course Descriptions PDF
College of Pharmacy
- Our Department
- DEI Efforts
- About the PhD Graduate Program
- PhD Requirements
- Research Facilities and Equipment
- Faculty Research Foci
- Postdocs & Research Staff
- Abul-Hajj and Hanna Award for Exceptional Graduate Student in Medicinal Chemistry
- Ole Gisvold Lecture
- Philip S. Portoghese Distinguished Lecture
- Taito O. Soine Lecture
- Distinguished Lectures
- Medicinal Chemistry Seminars
- Epigenetics Consortium Seminars
- Chemical Biology Initiative Seminars
- Epigenetics Consortium
- ACS Infectious Diseases
- From Digitalis to Ziagen
Admissions Process
- Before You Begin
- Prerequisites
- Application Checklist
- Students apply to our Medicinal Chemistry PhD program rather than to individual professors’ labs. The first semester of graduate school, students rotate through three different labs to experience the work in which our different professors are engaged in. After this, students are matched with professors depending on which professors have space/resources and mutual interest.
- While we look forward to reviewing your application, none of our professors are allowed or able to promise a spot in their particular lab should a student matriculate.
- We only accept applicants for fall term admission. Incomplete applications will not be reviewed. A complete application contains all required materials, as well as paying the Graduate School application fee. We only accept applicants into our Ph.D. program. Applications must be submitted by January 3rd.
- All applicants are considered for financial assistance and there are no additional forms needed beyond the application.
- We no longer require GRE scores. If submitted, they help further emphasize other areas of your application. Your application is not given an advantage for submitting GRE scores, and likewise, an application is not negatively impacted for not submitting GRE scores.
- For international students, please review the information regarding demonstration of English Proficiency as required by our Graduate School.
- The Medicinal Chemistry program welcomes applications from students with pharmacy, chemistry, or biology degrees, and students majoring in other degree programs that encompass chemical, biochemical, or biological fields of study are also encouraged to apply.
- A grade point average indicative of a strong undergraduate academic performance is required of all applicants.
- Students from non-English speaking countries must submit either a Test of English as a Foreign Language ( TOEFL ) score or an International English Language Testing System ( IELTS ) score. See Graduate School information about minimum performance levels . Competitive international applicants to the Medicinal Chemistry program have typically had TOEFL scores of 100 or higher or IELTS scores of 7 or higher.
- Please note that the University of Minnesota's institution code 6874 should be used for the TOEFL submissions.
- Please note that the Department of Medicinal Chemistry no longer requires GRE scores.
- A written statement explaining your interest in Medicinal Chemistry and why the graduate program at the University of Minnesota is a good fit for you.
- A brief summary of any prior work or research experience that has influenced your decision to pursue a PhD in Medicinal Chemistry with us.
- Your Resume or CV.
- Transcripts from all post-secondary institutions attended.
- Three letters of recommendations. You will be asked to provide contact information for your references, and they will be asked to submit their letter electronically through the online application system.
- An application fee ($75 domestic, $95 foreign students).
- Applications are only accepted for fall term admission. Incomplete applications will not be reviewed. You must fulfill all application requirements, including payment of the application fee, before your application is considered complete. Any inquiries about the application fee should be directed to the Graduate School.
- Applications for fall admission must be submitted by January 3rd.
Admissions FAQs
+ how does financial assistance work.
Students who have been accepted into the program with the assurance of financial assistance receive such support in the form of either teaching assistantships, research assistantships, or fellowships.
All students on assistantships and fellowships receive a tuition waiver and a comprehensive health care benefits package. Students do not need to fill out any additional forms when applying to be considered for financial support.
Students are fully supported throughout the 5 years of their studies as long as satisfactory progress is being made toward a degree.
+ Do prospective students visit campus?
Prospective students will be invited to our recruitment weekend. Here, you will have an opportunity to interact with faculty and students, tour our facilities, see the campus, and explore the Twin Cities.
Generally this occurs the first weekend in March.
+ What about student diversity?
The Department of Medicinal Chemistry is committed to fostering a diverse and inclusive learning environment that values and celebrates the unique perspectives, experiences, and backgrounds of all students. We recognize that diversity enriches our educational and research experiences, as it brings together individuals with unique viewpoints and life experiences.
Please visit the Graduate School Diversity Office for more information.
+ I still have questions - who do I contact?
Please contact us via email at: [email protected]
Department of Medicinal Chemistry 8-101 Weaver-Densford Hall 308 SE Harvard Street Minneapolis, MN 55455
- About Our College
- Message from the Dean
- Our Campuses Overview
- Duluth Campus
- Twin Cities Campus
- Living in MN
- Our Faculty
- Faculty by Department
- Administration
- Our Commitment to Equity, Diversity & Inclusion
- Job Opportunities
- Degrees and Programs Overview
- Doctor of Pharmacy
- Dual Degree Programs
- Graduate Programs
- Medical Laboratory Sciences Program
- Occupational Therapy Program
- Postgraduate Pharmacy Residency Program Overview
- Choosing the University of Minnesota
- Mission and Vision
- Comprehensive Medication Management
- Learning Experiences
- Program Administration
- Admissions Overview
- Brochures and Other Materials
- Recruitment Events
- Virtual Open House
- Residency Sites & Emphasis Areas
- Meet Our Current Residents
- News, Events and Publications
- Summer Undergraduate Experience Program
- Undergraduate Electives Overview
- Available Courses Overview
- PHAR 1001: Orientation to Pharmacy
- PHAR 1002: Medical Terminology
- PHAR 1003: Non-Prescription Medications and Self-Care
- PHAR 1004: Common Prescription Drugs and Diseases
- PHAR 2002: Precision Medicine and Health: Understanding the Personal Genome
- PHAR 3206: Foundations of Health Literacy
- PHAR 3700/5700: Fundamentals of Pharmacotherapy
- PHAR 4204W/5204: Drugs and the U.S. Healthcare System
- PHAR 5201: Applied Medical Terminology
- PHAR 5205: Obesity: Issues, Interventions, Innovations
- PHAR 5310: Topics in Pharmacy Ethics (Pandemics)
- Continuing Professional Development
- Departments & Divisions Overview
- Experimental and Clinical Pharmacology
- Medical Laboratory Sciences
- Medicinal Chemistry
- Occupational Therapy
- Office of Professional & Clinical Affairs
- Pharmaceutical Care & Health Systems
- Pharmaceutics
- Pharmacy Practice and Pharmaceutical Sciences
- Professional Education Division
- Centers and Institutes Overview
- Brain Barriers Research Center
- Center for Allied Health Programs
- Center for Clinical and Cognitive Neuropharmacology
- Center for Drug Design
- Center for Forecasting Drug Response
- Center for Leading Healthcare Change
- Center for Orphan Drug Research
- Epilepsy Research and Education Program
- The Institute for Therapeutics Discovery and Development
- Institute of Personalized Medicine (IPM)
- PRIME Institute
- Wulling Center for Innovation & Scholarship in Pharmacy Education
- Research Overview
- Research Service Labs
- Endowed Chairs/Professors
- Expertise Guide
- Office of the Associate Dean for Research
- Medical Laboratory Sciences Alumni
- Occupational Therapy Alumni
- Pharmacy Alumni
- Events and Awards
- Celebrating Our Alumni
- MNovationRX
- Covid-19 Stories
- Publications & Newsletters
- Mental Health First Aid Training
- Enroll & Pay
- KU School of Pharmacy
- Doctor of Pharmacy (Pharm.D.)
- Pharmaceutical Chemistry
- Pharmacology & Toxicology
- Pharmacy Practice
- Neuroscience
Doctor of Philosophy (Ph.D.) in Medicinal Chemistry
The KU Department of Medicinal Chemistry provides Ph.D. students a strong foundation in organic and medicinal chemistry with flexibility for additional emphasis in aspects of biochemistry, pharmacology and other biological sciences.
Apply for Ph.D.
Ph.D. Program Overview
Standard ku graduate admission requirements —.
Students must meet all requirements for Graduate Admissions .
Prerequisites —
Previous degree requirement —.
B.S. or M.S. degree in pharmacy, medicinal chemistry, chemistry, biochemistry, or a closely-related field
Grade Point Average (GPA) —
Applicants must have a minimum cumulative GPA of a 3.0 on a 4.0 scale.
Graduate Record Exam (GRE) Scores —
- GRE General Test is recommended but not required.
- GRE scores should be sent directly to the University of Kansas and to the Department of Medicinal Chemistry (codes: KU-6871, Medicinal Chemistry - 0621).
- Although not mandatory, applicants are encouraged to also take the subject test in chemistry.
English Proficiency Requirements —
Non-native English speakers must demonstrate proficiency in reading, writing and listening via English Proficiency Scores from the TOEFL, IELTS or PTE test. See KU's English Proficiency Requirements for detailed information, including minimum score requirements. Request that the testing agency send your official scores directly to KU (codes: KU-6871, Pharmacy-47).
Time to Complete —
The program typically takes five years to complete. Required core graduate courses for students who meet standard requirements can be completed within the first two years of study. Students attend year-round with time off for holidays and vacations.
Minimum Enrollment —
Students enroll in at least 9 credit hours in both the fall and spring semesters and 6 hours in the summer. Students must take all required courses, even if that requires more than the minimum hours a given term. Students must be enrolled in at least 1 hour of thesis or dissertation research each term (MDCM 895 or 999), regardless of other coursework.
Foundational Prerequisite Courses —
One year of organic chemistry with laboratory (equivalent to CHEM 624, 625, 626, 627) and at least one course in physical chemistry (equivalent to CHEM 640, 646) and one course biochemistry (see note below).
Note About Biochemistry A one-semester survey course in biochemistry is acceptable if the student received a grade of B or better in the course OR if the student scores a 70 or better on the ACS Biochemistry placement exam given to entering graduate students in the fall (one try only will be allowed). If neither of these applies, the student will take one semester of biochemistry through the Department of Medicinal Chemistry (MDCM 701).
See Courses - Ph.D. for details about required coursework, safety training and academic standing.
Research Requirements —
Graduate degrees in medicinal chemistry are research-based and awarded only after a student has made a significant, in-depth contribution of new knowledge to the field in the form of research publications and the M.S. Thesis or the Doctoral Dissertation.
Academic Standing —
At the end of the first semester, continuance in the program is dependent upon satisfactory academic program progress.
Comprehensive Written Examination —
After the spring semester of year one, students take a comprehensive written examination and must score 70% or higher. A score of 50%-69% qualifies students for one additional attempt, which must occur before fall semester of year two. A score below 50% will typically result in dismissal.
Comprehensive Oral Examination —
Students take a comprehensive oral exam after the first two years of coursework. Successful completion results in the student attaining the status of doctoral candidate. A non-thesis M.S. degree is automatically awarded to all students after the successful completion of their oral comprehensive examination.
Seminar Presentations —
Students must prepare and present two seminars in the departmental seminar series. The first is the Literature Seminar (MDCM 798) and presented during the spring semester of year two. The second seminar is the research seminar (MDCM 799), during the fall semester of year four and highlights research progress.
Original Research Proposal —
As part of the “Proposal Preparation” course (MDCM 980), during the fall semester of year three, students prepare an original proposal (NIH format), and submit it to the faculty for evaluation. This proposal is based on the same topic as their literature seminar.
Research Rotations —
Students perform two research rotations during the first semester and are assigned a research advisor, both for rotations and the final research group assignment. Assignments are based on student’s preference as well as the availability of funding and research space.
Student Self-Assessment —
Starting in the third year, students are required to complete a yearly self-assessment of their goals and progress toward those goals.
Dissertation Defense —
The final requirements for the Ph.D. degree are the preparation and defense of a dissertation based on the original laboratory research conducted by the student.
Safety Training —
Students must comply with training required by the KU Department of Environment, Health and Safety and the Department of Medicinal Chemistry. Training can include research seminars, hands-on training and online training. Safety training specific to assigned labs must also be completed before students are allowed to begin laboratory work.
Director of Graduate Studies Course Mark Farrell Assistant Professor [email protected] 785-864-1610
Graduate Student Recruiting Application [email protected] 785-864-4495
KU Graduate Admissions [email protected] 785-864-3140
- Faculty/Staff Access

Graduate Program
Ph.d. in medicinal chemistry and molecular pharmacology.

The Department of Medicinal Chemistry and Molecular Pharmacology (MCMP) is one of the top-rated programs in the country and is unique because it combines both medicinal chemistry and molecular pharmacology. Students in our PhD program will be trained in an environment that combines chemical and biological approaches, which is essential for translating basic discoveries into novel therapeutics. The scientific approaches taken by students and over 25 faculty in MCMP includes medicinal chemistry and chemical biology, cell and molecular biology, molecular pharmacology, biophysical and computational chemistry, systems biology, functional and pharmacogenomics. Biomedical research topics covered in MCMP can be broadly grouped in three general areas: cancer biology, neuroscience, and infectious diseases.
Our Ph.D. program is fully committed to excellence and innovation in the education of Ph.D. students. We are an inclusive and supportive environment, filled with energetic and creative students, fellows, faculty, and staff. Explore our faculty pages and contact us if you are interested in graduate studies and our Ph.D. program.
The MCMP Graduate Program
- Multidisciplinary Drug Discovery
- Molecular Pharmacology
- Biophysical and Computational Chemistry
- Functional Genomics and Pharmacogenomics
- Medicinal Chemistry and Chemical Biology
- Structural Biology
- Systems Biology
- #7 Pharmacy and #5 Most innovative - US News & World report 2020
- Top 10 Pharmacology in USA - QS rankings 2019
- Top 10 US Public University - The Wall Street Journal, 2020
- 5 consecutive years of Purdue University record research funding
- #13 Worldwide for US Patents - National Academy of Inventors, 2020
We have a world-class reputation for equipping young investigators with the skills andknowledge needed to be successful researchers in the pharmaceutical sciences. Graduate students play an integral role in accomplishing our research mission, and the rich scientific environment that exists in the department can be largely attributed to the outstanding quality of students admitted to the Ph.D. program. Indeed, a number of graduate students in the department have received extramural awards from agencies such as NIH, NSF, and the PhRMA Foundation.
Only students seeking a Ph.D. are admitted to the MCMP graduate program. Two-thirds of the graduate students in MCMP are admitted directly to the departmental graduate program, whereas the other third choose MCMP as their academic home after having entered graduate school through campus interdisciplinary training programs. There are currently over 80 graduate students enrolled in the department, the vast majority of whom are engaged in studies leading to the Ph.D. degree. The presence of approximately 30 postdoctoral associates and other research staff professionals further enriches the intellectual atmosphere.
Information on the Program
- Tabular time line of the program
- Policies and Regulations
- Detailed information on the application and admission processes
- Graduate students in the department are fully supported through various mechanisms including fellowships, research assistantships, and teaching assistantships. Further information on financing is available.
Graduate Program Information from the College of Pharmacy and the University
- Policies and Regulations Manual for Graduate Programs
- Graduate Pharmacy Programs - Useful information about graduate study in the College
- Graduate Pharmacy Program Admissions - Useful information and FAQ about admissions and application for graduate programs in the College
Need more information? Feel free to call us at 765-494-1362 or send an inquiry to [email protected] .
PhD Program: Medicinal Chemistry
About the track.
The medicinal chemistry track encompasses drug discovery and prepares you with the means to study the behavior of chemical substances at the molecular level.
- You will use computational, biochemical and cell-based screening technologies to identify natural and synthetic compounds with pharmacological activity.
- You will study structure-activity relationships to understand the mechanisms of drug action.
- Your research will be directed towards the identification, synthesis and development of new chemical molecules suitable for biological studies and eventually therapeutic use.
Download our brochure
Faculty Associated with this Track
- Donna Huryn, PhD
- Paul Johnston, PhD
- Velvet Journigan, PhD
- Jaden Jun, PhD
- Terance McGuire, PhD
- Peter Wipf, PhD
- Xiangqun Xie, PhD, MBA
- Wei Zhang, PhD
Download the competency requirements for a PhD in Pharmaceutical Sciences
Primary Contact
Xiangqun (Sean) Xie , PhD, MBA Professor 206 Salk Pavilion 412-383-5276 [email protected]
- Prospective Student Inquiries [email protected]
- General Inquiries [email protected]
- Job Posting Request
- Building Hours & Maps
- Prospective Students
- Current Students
What's Happening
Student resources, quick links.
- Alumni & Friends
Update Your Information
- Academic Departments
- Clinical Pharmacy
- Medicinal Chemistry
- Pharmaceutical Sciences
Research in Clinical Pharmacy
Research in medicinal chemistry, research in pharmaceutical sciences, research cores and services, biointerfaces institute, michigan drug discovery.
- Michigan Institute for Clinical & Health Research
Translational Oncology Program
- Faculty Publications
- Research Opportunities
- Research Collaborations
UM Pharmacy Professor Research Outreach (PRO)
- About the College
Message from the Dean
- Dean Search
Accreditation
New cop building, our history, our leadership.
- Our Mission, Vision & Organization
Teaching Excellence Awards
Job openings.
- Diversity, Equity & Inclusion
Department Contact Info
Emergency information.
- COVID-19 Updates
Sexual and Gender-Based Misconduct
- Alternative and Complementary Medicines
- Diagnosis and Health Conditions
- Healthy Choices
- Information for Caregivers
- Medication Information
- Other Resources
- COP Directory
Search form
- Alumni & Friends
Why U-M Pharmacy?
Recruitment events, student blogs, career potential, program overview.

- Information Request Form
PharmD Program
- Experiential Education
- Pharmacy Phamilies
- PharmD Curriculum
- Assessments
- Pharmacy Student Ambassadors
- Pre-Pharmacy Student Organization (PPSO)
PhD in Clinical Pharmacy
Phd in medicinal chemistry, phd in pharmaceutical sciences, ms in integrated pharmaceutical sciences.
- Career Flexibility
BS in Pharmaceutical Sciences
- Program Goals
- Fast Track to PharmD
- Student Services
- Research and Honors Program
- Student Outcomes
Dual Programs
- Dual PharmD and MBA Program
- Dual PharmD and MPH Program
- Dual PharmD and PhD Program
Post-Doc in Clinical Pharmacy
Residency program, ambulatory care enrichment program, research experiences for undergraduates program.
- Eligibility
- Program Schedule
- Application
- Pharmacy Scholars Program
- Frequently Asked Questions
- Income Guidelines
Assignment of Credit Hours
Pharmacy community college connect, postdoctoral collegiate fellows program.
- Expectations of Faculty Mentors
- Faculty Mentor List
- Review & Selection
PharmD Program Admissions
- Application Overview
- PharmD Prerequisites
- Preferred Admission Programs
- Applicant Characteristics
PhD Program Admissions
Ms program admissions, bachelors program admissions, funding your education, financial aid brochure.
- Tuition and Fees
- PharmD Scholarships
- Graduate Support
Student Organizations
- Student News
Course Descriptions
Student affairs.
- Financial Aid & Scholarship
- Advising & Registration
- Career Counseling
- Personal Counseling
- Campus Resources
- Student Affairs Directory
Career Connections
Student handbook, access pharmacy, campus groups, cornerstone learning, outlook in the cloud, rx preceptor, taubman library, wolverine access.
- Prescott & Emeritus Celebration
Alumni Awards
- Alumni News
Board of Governors
- Nomination Form
- Board Member Position Description
Job Opportunities
Submit personal news, meet the advancement team, update your alumni record.
- News & Events
- Previous Faculty Spotlights
- Department Metrics
- Research Laboratories
- CPTS PhD Program
- CPTS Fellowship Program
- Post-Graduate Residency
- REACH Fellowship
- Infectious Diseases Fellowship
- ACE Program
- Vision and Mission
- Department Directory
- Simulated Patient Program
- Biochemical NMR Core
- Clinical Pharmacogenomics Laboratory
- Pharmacokinetic and Mass Spectrometry Core
- Vahlteich Medicinal Chemistry Core
Michigan Institute for Clinical & Health Research
- Previous Seminars
- Newly Awarded
- Available Funding
- Grant Tools "Coming Soon"
- Giving Tuesday
- Faculty News
- Research News
- Annual Report Archive
- Upcoming Events
- Newsletter Archive
Our Mission, Vision & Organization
- COP Organization Chart
- COP Strategic Plan
Diversity, Equity & Inclusion
- Dean's Vision Statement
- DEI Strategic Plan
- Upcoming News & Events
- REU Program
- Education & Training
- McKesson Foundation Health Equity Speaker Series
- Resources & Support
- Concern Reporting
- Emergency FAQs
- Stay Informed!
MedChem Banner

TOP RESEARCH EXPENDITURE IN THE U.S.
Medicinal chemistry involves the application of a number of specialized disciplinary approaches all focused on the ultimate goal of drug discovery. Drug target identification and validation, rational (target-based) drug design, structural biology, computational-based drug design, methods development (chemical, biochemical, and computational), and “Hit-to-lead” development are all aspects of medicinal chemistry. The techniques and approaches of chemical biology, synthetic organic chemistry, combinatorial (bio)chemistry, mechanistic enzymology, computational chemistry, chemical genomics, and high-throughput screening are all used applied by medicinal chemists towards drug discovery.
For our Pharm.D. students, medicinal chemistry is integrated with pharmacology to present a coherent picture of the principles of drug action. Pharmacology mainly deals with drug action at the cellular, tissue/organ and organism levels. Medicinal chemistry focuses on the molecular aspects of drug action: interactions with the drug targets from both the drug and the target point of view, the relationship of drug chemical structure to drug action and the effects of metabolism on the drug structure and hence its action.
In 1913, the University of Michigan first outlined a graduate program offering M.A., M.S., Ph.D. and D.Sc. degrees in a joint effort between the Graduate Department (now the Horace H. Rackham School of Graduate Studies) and the Pharmacy Department (now College of Pharmacy). As a discipline, Medicinal Chemistry in the United States started with the appointment of Dr. F. F. Blicke as Assistant Professor of Pharmaceutical Chemistry in 1926. Prof. Blicke initiated the first graduate education program in Pharmaceutical Chemistry, focusing on synthetic organic chemistry. The program expanded in the 1950s to include analytical aspects and pharmaceutics. After Prof. Blicke’s retirement in 1960, his former student, Prof. J. H. Burkhalter returned to the College and argued for an independent graduate education program in Medicinal Chemistry. Along with the support of Graduate School Dean Alfred Sussman and the participation of a core group of interdepartmental faculty (within and outside of the College of Pharmacy), in 1967 Prof. Burkhalter established the Interdepartmental Program in Medicinal Chemistry (Med Chem IDP). The Med Chem IDP was established to train students in a broad range of chemically-based disciplines so that its graduates are able to apply the rigor and methods of the physical sciences to drug discovery research. Subsequently, in 1973 Prof. Raymond Counsell and in 1977 Prof. Leroy B. Townsend were appointed as Director of the Med Chem IDP. The Med Chem IDP is administered by the Horace H. Rackham School of Graduate Studies with direct oversight by the College of Pharmacy.
In 1999, in response to the significant growth of the College of Pharmacy under previous Dean Ara G. Paul, then Dean George L. Kenyon initiated a process of departmentalization of the College of Pharmacy. Prof. James K. Coward was the first Chair of the Department of Medicinal Chemistry and Director of the Med Chem IDP. The Department of Medicinal Chemistry is the administrative component of the College of Pharmacy that oversees the Medicinal Chemistry faculty, research scientists and postdoctoral fellows (e.g., recruitment, mentoring, evaluation), has responsibility for the medicinal chemistry Pharm.D. and Ph.D. courses and seminar program, and coordinates the participation of medicinal chemistry faculty in College-level committees and other administrative duties.
The Department of Medicinal Chemistry is the home for the Med Chem IDP Ph.D. program, which serves to administer the Med Chem Ph.D. program, with responsibility for graduate student recruitment, training/mentoring, progression and graduation. The Med Chem IDP includes all faculty from the Department of Medicinal Chemistry as well as select faculty from the Department of Pharmaceutical Sciences in the College of Pharmacy and a variety of schools (e.g., Literature, Science and the Arts, Medical School) and departments at Michigan (e.g., Biological Chemistry, Biophysics, Chemistry, Pathology, Pharmacology, Radiology). Approximately half of the Med Chem IDP faculty have their primary appointments outside of the Department of Medicinal Chemistry. These faculty currently mentor ~20% of the Med Chem Ph.D. students and are fully engaged in the Med Chem Ph.D. program in many other ways including seminar attendance, recruitment of students, teaching in our graduate courses, and serving on candidacy and dissertation committees. There is an annual meeting of the Med Chem IDP faculty to review the status of the IDP and the students.

Graduate Programs
Explore information.
If you want a career at the forefront of new drug development, there's nowhere better to study in Ohio than The University of Toledo.
UToledo’s College of Pharmacy and Pharmaceutical Sciences has been ranked first in Ohio and eighth in the U.S. for teaching and value. UToledo also is ranked among the nation's best pharmacy schools by U.S. News & World Report.
Our master’s and doctoral programs in Medicinal Chemistry focus on the theory and practice of drug design. Our strength — and what sets us apart from other programs — is our focus on chemistry and advanced biology. Not many universities emphasize both.
Graduate students learn biological techniques to identify targets. They use chemistry to design drugs to affect those targets. This holistic, interdisciplinary approach makes our graduates more marketable.
Employers also love that our Medicinal Chemistry students are prepared for hands-on research. UToledo graduates of the Medicinal Chemistry Ph.D. program go on to prestigious post-doctoral appointments and high-level jobs in the pharmaceutical industry or academia. M.S. graduates go on to Ph.D. programs at UToledo or other universities or directly into industry.
Pharm.D./Ph.D. Dual Degree Program
In addition to our Ph.D. and M.S. programs in Medicinal Chemistry, we also offer a dual degree program for those who want to earn both their Ph.D. and Pharm.D. degrees.
Top Reasons to Study medicinal chemistry at UToledo
- Small classes. Our classes are offered in a small-group tutorial format. You have extensive, direct access to faculty mentors.
- Intensive research. Begin your doctoral program rotating through at least two faculty laboratories, where you will conduct small research projects. These rotations will inform and inspire your future dissertation work. M.S. students enter a lab during their second semesters.
- Internships. Participate in an optional internship with a Toledo, Ohio-based company or one of our international partners. These experiences have turned into career-track jobs for many Medicinal Chemistry graduates.
- Health Science Campus. Our college's location on UToledo's Health Science Campus allows graduate students to collaborate with students in other healthcare professions and access research labs, pharmacies and more. All students also have access to the chemical instrumentation on Main Campus.
- The Shimadzu Laboratory for Pharmaceutical Research Excellence has the latest advanced instruments, including a triple-quadrupole mass spectrometer. The instruments allow M.S. and Ph.D. students to research metabolism, disease biomarkers, DNA damage and other areas.
- The Center for Drug Design and Development is a university-based hub for collaborative research with the pharmaceutical industry. The UToledo Department of Medicinal and Biological Chemistry works closely with the center.
- Financial support. Students in the Ph.D. program are often supported throughout the program as teaching assistants or research assistants with funding from research grants. Assistantships generally require teaching and research.
Graduate students, postdoctoral fellows, research assistants and visiting scholars contribute to a vital research environment at UToledo.
UToledo faculty in the Department of Medicinal and Biological Chemistry are skilled researchers and teachers. They are recognized authorities in their areas of specialization and conduct research that contributes to the development of new treatments, practices and innovations.
Master’s and doctoral students are trained in applied research in rational drug design. They work closely on research with faculty members for their theses.
UToledo Medicinal Chemists collaborate with chemistry, biology and medical faculty on research. They are involved in research in:
- Neuroscience
- Cancer therapy and vaccines
- Kidney and cardiovascular diseases
- Organic synthesis
- Autoimmunology and basic immunology
- Inflammation and obesity
- Targeted drug design and development
What jobs can I get with a medicinal chemistry degree?
Graduates of UToledo's Medicinal Chemistry master's and Ph.D. programs have had a 100% job placement rate during the past few years. The interdisciplinary training our graduates receive is in demand in industry and academia.
Employers include:
- Pharmaceutical companies
- Biotechnology companies
- Hospital and government laboratories
- Universities
Non-laboratory opportunities include:
- Clinical trial administration
- Scientific writing
- Intellectual property
- University faculty and other positions in academia
Our graduates have been accepted into doctoral programs and offered postdoctoral fellowships at:
- Walter Reed Army Institute of Research
- Harvard University
- Columbia University
- Ohio State University
- University of Michigan
- University of Georgia
- Johns Hopkins University
- University of Chicago
- Northwestern University
- Scripps Research Institute, Florida
Our graduates in Medicinal Chemistry have been employed in the following positions:
- Chemist, Anatrace, Maumee, Ohio
- Chemist, Stepan Company, Chicago
- Quality control technician, Mondelez International, Deerfield, Ill.
- Portfolio manager, medical imaging, U.S. Department of Defense
- Medical director/compliance officer, Informed Medical Communications, Rockville, Md.
- Research analyst, Compass Labs, Memphis, Tenn.
- Medical writer, Integra Lifesciences, Plainsboro, N.J.
- Associate professor, Thomas J. Long School of Pharmacy & Health Sciences, Department of Pharmaceutics & Medicinal Chemistry, Stockton, Calif.
- Director general, Arab Company for Drug Industries and Medical Appliances, and assistant professor, University of Jordan, Amman, Jordan
- Program manager for peer-reviewed Alzheimer’s/epilepsy research programs, U.S. Department of Defense
How to Apply to Graduate School
Find your next steps whether you are a new student, readmit student or guest student.
Back to Top
Doctor of Philosophy in Medicinal Chemistry (PhD)
Location: Boston Start Term: Fall Either build on your master’s-level knowledge or start directly on your PhD examining the behavior of chemical substances at the molecular level and conducting research related to the development of new drugs and novel targets.
Research Drug Design and Synthesis in Some of the World’s Most Advanced Laboratories
Your doctoral journey.
Our research-and-development focused curriculum prepares you for roles at the frontier of drug discovery and enhancement in the pharmaceutical industry. You’ll work side-by-side with globally respected faculty working on novel drug targets and diseases. This program is STEM-designated , qualifying international students for an additional two years of OPT work in the United States.
First-Year Experience (following completion of your MS degree)
- deepen your grasp of pharmaceutical science principles
- study medicinal, organic, and bio-organic chemistry and spectroscopic analysis
- participate in related laboratory rotations and graduate seminars
Second-Year Experience
- engage in research in your desired area of specialization
- continue to participate in related graduate seminars
Third-Year Experience
- develop a thorough understanding of drug metabolism
- extend your research in a desired area of specialization and create a grant proposal
Medicinal Chemist compensation reported by Payscale.com in 2022.
The projected professional growth between 2022 and 2032 is 6%, faster than average. (Bureau of Labor Statistics)
Massachusetts is #1 in Industry investment in R&D per capita. (Massachusetts Life Sciences Center)
Developing Future Leaders in the Pharmaceutical Sciences
Dream Jobs That Help Humanity
As a medicinal chemist, you’ll work with some of the most dynamic technologies and life-saving breakthroughs in healthcare.
Working at the Frontier of Therapeutics
Jose Mauricio Paredes Quiroz, MS ’22, pursues his calling as a drug hunter working on RNA-based therapeutics at Dicerna Pharmaceuticals.
Students Publish with Pharmaceutical Sciences Professor
Professor Swati Betharia, PhD in Pharmacology ’11, led a research team of undergraduate and graduate students to a key finding on neuroprotection against metal toxicity.
Graduate Degrees in Pharmaceutical Sciences
As well as the PhD in Medicinal Chemistry, we offer five additional opportunities for graduate study at the master’s, doctoral, and certificate levels.
Master of Science in Medicinal Chemistry
Two-year, full-time program on the Boston campus.
Master of Science in Pharmaceutics
Two-year, full-time program on the Boston and Worcester campuses.
Master of Science in Clinical Research
Flexible full-time or part-time program on the Boston campus and online.
Graduate Certificate in Clinical Research
Flexible three-course program on the Boston campus and online.
Doctor of Philosophy in Pharmaceutics
50-credit, full-time program on the Boston and Worcester campuses.
Download a Program Fact Sheet
Download a program fact sheet for a snapshot of the Doctor of Philosophy in Medicinal Chemistry.
Cookie Notice
Your browser is unsupported
We recommend using the latest version of IE11, Edge, Chrome, Firefox or Safari.
College of Pharmacy - Chicago | Rockford
Phd in pharmaceutical sciences.
We enable students with backgrounds in fundamental sciences to become leaders in pharmaceutical sciences
Located in the vibrant and multicultural city of Chicago, UIC's PhD Program in Pharmaceutical Sciences is one of the strongest and largest of its type in the United States. Our college is consistently ranked in the top ten in terms of funds secured annually from the National Institutes of Health and by US News and World Report. We pride ourselves on giving students from all types of backgrounds the tools they need to become independent researchers. Students in the program select one of the program concentrations, described below.
Important dates Heading link Copy link

We are so pleased you are considering graduate studies in Pharmaceutical Sciences at the University of Illinois Chicago! Although Pharmaceutical Sciences is one of the best graduate programs of its kind in the country, our real pride is mentoring students into independent researchers who become leaders in our field. The program has some unique strengths, including providing flexibility to carry out internships in your later years. Have a look around our website. If you have questions, feel free to reach out to us at [email protected] . We look forward to reading your application! Debra Tonetti, PhD | Professor, Pharmaceutical Sciences
Program Coursework Heading link Copy link
All students in the Pharmaceutical Sciences program take the following courses. Additional concentration coursework is also required and is shown in each of the concentration tabs.
- Drug Discovery, Design, and Development (PSCI 501, 3 credit hours)
- Training in Research Presentation (PSCI 502, 1 credit hour)
- PSCI 503: Biostatistics for Pharmaceutical Scientists (1 credit hour)
- BSTT 400: Biostatistics I (4 credit hours) [Note: BSTT 400 is required for the Pharmaceutics and Drug Delivery concentration]
- Scientific Ethics and the Responsible Conduct of Research (GC 501, 1 credit hour)
- Research Rotation (PSCI 592; 3-4 credit hours)
- PSCI PhD Course Requirements
- PSCI Department Course Descriptions
Program Concentrations Heading link Copy link
Five concentrations comprise the PhD program in Pharmaceutical Sciences. Click on the tabs below to learn more about each of them. To see the faculty mentors for each concentration, visit the Faculty Mentors page .
Chemistry in Drug Discovery
Concentration description.
Faculty in the Chemistry in Drug Discovery concentration use the tools and techniques of chemistry to discover and develop new chemical probes and potential therapeutics. Students in this concentration learn how to design, synthesize, characterize and analyze small molecules, peptides, and proteins.
Concentration Coursework
Students in the Chemistry in Drug Discovery Concentration take the following courses:
- Fundamental of Drug Action I (PHAR 422, 4 credit hours)
- Principles of Medicinal Chemistry (PSCI 530, 5 credit hours)
- Electives (9 credit hours)
Concentration Coordinator
Prof. Terry Moore ([email protected])
Molecular Mechanisms and Therapeutics
The Molecular Mechanisms and Therapeutics concentration is designed to provide advanced understanding of fundamental causes of diseases, strategies that identify new drug targets, and mechanistic explanations of how drugs work (or fail) from the perspective of the target and systems they impact. Faculty affiliated with MMT integrate a wide variety of molecular, biochemical, genetic, bioinformatic, and bioengineering approaches to study mechanisms of pathogenesis ranging from infectious diseases to cancer. Students will enroll in fundamental molecular and cellular biology courses and select elective courses in areas of their focused research.
Students in the Molecular Mechanisms and Therapeutics Concentration take the following courses:
- Biochemistry (e.g., GEMS 501 or equivalent graduate-level biochemistry course, 3 credit hours)
- Molecular Biology (e.g., GEMS 502 or equivalent molecular biology course, 3 credit hours)
- Biostatistics I (BSTT 400, 4 credit hours)
- Molecular Genetics (GEMS 511, 3 credit hours)
- Receptor Pharmacology and Cell Signaling (GEMS 515, 3 credit hours)
- Microbial Pathogenesis (MIM 560, 3 credit hours)
- Cancer Biology and Therapeutics (PSCI 540, 3 credit hours)
Prof. Alessandra Eustaquio ( [email protected] )
Pharmaceutics and Drug Delivery
Faculty in the Pharmaceutics and Drug Delivery concentration use the tools and techniques of physical and biologic sciences and engineering to understand and develop delivery systems and formulations for therapeutic molecules and control the biodistribution of therapeutic molecules. Students in this concentration learn how to design, synthesize, characterize and analyze novel materials and drug delivery systems and design and develop technologies related to therapeutic distribution in the body.
Students in the Pharmaceutics and Drug Delivery Concentration take the following courses:
- *This 4 credit hour course will count 1 hour toward the program core statistics requirement and 3 hours toward the Pharmaceutics and Drug Delivery concentration requirements. Students will not receive credit for two introductory statistics courses.
- Essentials for Animal Research (GC 470, 1 credit hour)
- Experimental Animal Techniques (GC 471, 2 credit hours)
- Principles of Pharmaceutics and Drug Delivery (PSCI 510, 3 credit hours)
Prof. Richard Gemeinhart ([email protected])
Pharmacognosy
Faculty research programs in the Pharmacognosy concentration aim to develop therapeutics from natural products and to study the mechanisms of pain, cancers, and a wide array of infectious and tropical diseases. Students of this concentration are trained in a combination of bioinformatics, synthetic biology, genetic engineering, chromatography, and spectroscopy to achieve these goals.
Students in the Pharmacognosy Concentration take the following courses:
- Research Techniques in Pharmacognosy (PSCI 520 or equivalent; 3 credit hours)
- Structure Elucidation of Natural Products (PSCI 521 or equivalent; 3 credit hours)
- Advanced Pharmacognosy (PSCI 522 or equivalent; 3 credit hours)
Prof. Brian Murphy ([email protected])
PharmD/PhD Joint Program Heading link Copy link
Pharmaceutical Sciences participates in the joint PharmD/PhD program, which trains students for careers in academic pharmacy and bench science research. Students admitted to this joint program participate in the PharmD curriculum and pursue original doctoral research projects in the laboratories of the university’s graduate faculty in the Department of Pharmaceutical Sciences.
The joint program offers the potential of reducing the time of earning both degrees in sequence (9 or more years) by approximately two years. The trade-off is that both degrees are awarded at the end of the training period and neither degree can be received before the other is completed.
The PharmD/PhD program is for exceptional, highly motivated and achieving students ready to meet the challenge of increased academic load and independent research project.
Program coordinator: Dr. Lindsey McQuade ( [email protected] )
- Joint PharmD/PhD Course Requirements
- Joint PharmD/PhD Program Page
PSCI Slideshow Heading link Copy link
- Go to slide 1
- Go to slide 2
- Go to slide 3
- Go to slide 4
- Go to slide 5
- Go to slide 6
- Go to slide 7
- Go to slide 8
- Go to slide 9

- Go to the next slide
- Go to the previous slide

Pride Points for PhD PSCI Heading link Copy link
$ 35,162 annual graduate stipend for students on teaching assistantship or research assistantship
33 internships completed by department graduate students in the last five years
19 students currently on training grant or fellowship
# 7 nationally ranked College of Pharmacy according to US News
# 7 nationally ranked total research funding among Colleges of Pharmacy according to AACP

Start your application Heading link Copy link
The Pharmaceutical Sciences Program at UIC offers a supportive, inclusive environment and rigorous academic preparation for students who are interested in careers in pharmaceutical sciences. If you have any questions about the program or about your application, please contact [email protected].
Get in touch: Contact Us
PhD Program

Professor Wender discusses chemistry with his graduate students.
Doctoral study in chemistry at Stanford University prepares students for research and teaching careers with diverse emphases in basic, life, medical, physical, energy, materials, and environmental sciences.
The Department of Chemistry offers opportunities for graduate study spanning contemporary subfields, including theoretical, organic, inorganic, physical, biophysical and biomedical chemistry and more. Much of the research defies easy classification along traditional divisions; cross-disciplinary collaborations with Stanford's many vibrant research departments and institutes is among factors distinguishing this world-class graduate program.
The Department of Chemistry is committed to providing academic advising in support of graduate student scholarly and professional development. This advising relationship entails collaborative and sustained engagement with mutual respect by both the adviser and advisee.
- The adviser is expected to meet at least monthly with the graduate student to discuss on-going research.
- There should be a yearly independent development plan (IDP) meeting between the graduate student and adviser. Topics include research progress, expectations for completion of PhD, areas for both the student and adviser to improve in their joint research effort.
- A research adviser should provide timely feedback on manuscripts and thesis chapters.
- Graduate students are active contributors to the advising relationship, proactively seeking academic and professional guidance and taking responsibility for informing themselves of policies and degree requirements for their graduate program.
- If there is a significant issue concerning the graduate student’s progress in research, the adviser must communicate this to the student and to the Graduate Studies Committee in writing. This feedback should include the issues, what needs to be done to overcome these issues and by when.
Academic advising by Stanford faculty is a critical component of all graduate students' education and additional resources can be found in the Policies and Best Practices for Advising Relationships at Stanford and the Guidelines for Faculty-Student Advising at Stanford .
Learn more about the program through the links below, and by exploring the research interests of the Chemistry Faculty and Courtesy Faculty .
Bouvé College of Health Sciences
Medicinal chemistry and drug discovery.
The Master of Science in Medicinal Chemistry and Drug Discovery integrates aspects of contemporary medicinal chemistry and pharmacology, emphasizing topics most relevant to therapeutics design, discovery, and action.
The Master of Science in Medicinal Chemistry and Drug Discovery offered by the Department of Pharmaceutical Sciences, develops students' knowledge in the design, synthesis, and mechanisms of action of novel biologically active compounds using modern biochemical, biophysical, and pharmacological approaches. The core curriculum focuses on a combination of synthetic organic chemistry, bioorganic chemistry, analytical chemistry, and pharmacology courses. Through in-depth elective courses, the program offers students the opportunity to develop medicinal chemistry expertise that can be applied to a practice-oriented career in the pharmaceutical industry. Graduates of the program will also be well prepared to enter related PhD programs at the university.
More Details
Unique features.
- The University is strategically located within the Boston-area biomedical and pharmaceutical/biotech-industry ecosystem
- The MS program provides opportunities for original research experience in department labs, and the option to do a literature or laboratory-based master's thesis for credit
- The interdisciplinary and comprehensive program is structured with core and elective courses using laboratory and computer-based technologies
- Northeastern’s School of Pharmacy and Pharmaceutical Sciences is consistently ranked first in NIH funding among all private U.S. schools of pharmacy.
- The program offers multiple opportunities for experiential learning through departmental and off campus internships and co-ops
- Program faculty includes industry-practiced scientists in drug discovery
- Those already employed in pharmaceutical/biotechnology careers can take specific courses for advanced training
Program Objectives
Upon completion of the MS in Medicinal Chemistry and Drug Discovery program, students will be able to:
- Explain current trends in organic synthesis as applied to therapeutics invention.
- Produce literature reviews encompassing retrieval, critical analysis, assessment, and written/oral presentation of current topics in drug discovery.
- Use organic-synthesis methodology and instrumentation in drug discovery.
- Apply experimental techniques to design, synthesize, and profile novel drug-like chemical entities.
- Maintain and use data, records, and notes/documentation according to ethical standards of research practice and academic integrity.
- Conceptualize and communicate to diverse constituencies in good oral and written English the principal concepts in medicinal chemistry as applied to drug discovery/development.
- Judge how social, economic, and ethical issues may impact drug discovery.
- Determine shortcomings of extant pharmacotherapeutics and how they may be overcome.
- Predict future trends in medicinal chemistry and drug discovery for synthesis of druggable chemical matter.
- Apply organic chemistry to advance drug discovery and development.
- Determine therapeutic areas where synthesis of new drug-like molecules is needed to satisfy unmet medical needs.
Career Outlook
Graduates of the Medicinal Chemistry and Drug Discovery program often work in chemistry labs synthesizing new chemical matter as potential drugs. Some graduates from the department have established careers as independent:
- Industry scientists and administrators (Big Pharma, biotech)
- Clinical laboratory staff
- Academic biomedical researchers
- Science teaching faculty
- Medical liaison specialists
- Pharmaceutical product representatives
- Medical writers
Looking for something different?
A graduate degree or certificate from Northeastern—a top-ranked university—can accelerate your career through rigorous academic coursework and hands-on professional experience in the area of your interest. Apply now—and take your career to the next level.
Program Costs
Finance Your Education We offer a variety of resources, including scholarships and assistantships.
How to Apply Learn more about the application process and requirements.
Requirements
- Application
- Application fee
- Two letters of recommendation
- Transcripts from all institutions attended
- Personal statement
- TOEFL required for applicants who do not hold a degree from a U.S. institution and whose native language is not English.
- GRE scores are optional
- Mathematics (including calculus)
- Biochemistry
- Organic chemistry
Please note: All international applicants will need to provide a WES
Are You an International Student? Find out what additional documents are required to apply.
Admissions Details Learn more about the Bouvé College of Health Sciences admissions process, policies, and required materials.
Admissions Dates
Fall only: June 1
Industry-aligned courses for in-demand careers.
For 100+ years, we’ve designed our programs with one thing in mind—your success. Explore the current program requirements and course descriptions, all designed to meet today’s industry needs and must-have skills.
View curriculum
Northeastern's signature experience-powered learning model has been at the heart of the university for more than a century. It combines world-class academics with professional practice, allowing you to acquire relevant, real-world skills you can immediately put into action in your current workplace. This makes a Northeastern education a dynamic, transformative experience, giving you countless opportunities to grow as a professional and person.
Our Faculty
Northeastern University faculty represents a broad cross-section of professional practices and fields, including finance, education, biomedical science, management, and the U.S. military. They serve as mentors and advisors and collaborate alongside you to solve the most pressing global challenges facing established and emerging markets.
You can see the faculty associated with the School of Pharmacy and Pharmaceutical Sciences programs here .
By enrolling in Northeastern, you’ll gain access to students at 13 campus locations, 300,000+ alumni, and 3,000 employer partners worldwide. Our global university system provides students unique opportunities to think locally and act globally while serving as a platform for scaling ideas, talent, and solutions.
Below is a look at where some of our alumni work, the positions they hold, and the skills they bring to their organization.
Where They Work
- Novartis Institutes for Biomedical Research
What They Do
- Healthcare Services
- Business Development
- Sales/Marketing
- Strategic Planning
- Competitive Surveillance
- Medical Liaison Specialists
- Pharmaceutical Product Representatives
- Medical Writers
What They're Skilled At
- Pharmaceutical Industry Practice
- Biotechnology
- Experimental Techniques in: Applied Pharmacology, Medicinal Chemistry and Human Biology
- Drug Design, Delivery, Action and Therapeutic Application
Learn more about Northeastern Alumni on Linkedin .
Related Articles

5 Alternative Careers For Psychology Majors that Aren’t Counseling

5 Research Careers With a Master’s in Psychology

Is Getting a DPT Worth It?
- Partnerships
Medicinal Chemistry and Drug Discovery (MS)
YOU ARE BOUVÉ

Learn to master therapeutics design and discovery.
The Medicinal Chemistry and Drug Discovery MS Program integrates aspects of contemporary medicinal chemistry and pharmacology, emphasizing topics most relevant to therapeutics design, discovery, and action.

The core curriculum focuses on a combination of synthetic organic chemistry, bioorganic chemistry, analytical chemistry, and pharmacology courses. Specialized, in-depth electives are offered in these areas.
The program develops students’ knowledge of medicinal chemistry through design, synthesis, pharmacological, and profiling of novel pharmacotherapeutics as applied to helping solve unmet medical needs.
For this purpose, many program graduates have established research careers in the pharmaceutical/biotech industry. Undergraduate prerequisites are general chemistry, organic chemistry, and biochemistry or cell/molecular biology.
Degree type: – MS in Medicinal Chemistry and Drug Discovery Study options: – On ground (Boston Campus) – Fall semester admissions only – *Full-time or part-time
Application deadlines: Jun 1
GRE: Optional
F1 Eligible: Yes
*International students must be full-time only
MS in Medical Chemistry and Drug Discovery
All Master’s programs in the Department of Pharmaceutical Sciences require a set of core courses taken by every MS student, regardless of program.
In addition, students in each program are required to take a defined set of discipline-specific courses and several general electives. The number of specialized and elective courses differs somewhat among programs. Students are expected to maintain a GPA of 3.000 (B) or higher in all coursework and cover 33 total semester hours.
The MS degree may be completed on either a full-time or part-time basis and may include an optional research thesis.
The curriculum includes opportunity for laboratory coursework and instruction in experimental design and data analysis.
International students are required to attend the program on a full-time basis. Students are expected to complete the degree requirements within two years if enrolled on a full-time basis, or within three to five years if enrolled on a part-time basis.
Internships provide an experiential component of the graduate curriculum that fosters professional development through work in industry and hospitals. In order to participate in an internship, students must:
- Complete two semesters with a grade-point average of 3.200 or better
- Be in good academic and professional standing (i.e., have no Professionalism Concern forms filed)
- Have no instances of academic dishonesty or blocks on enrollment.
Learn more about internships
Sample Curriculum
Sample curriculum, subject to change.
The chart below describes a sequence of courses as taught in the recent past. This chart is not a planning document for what course will be taught in which semester (spring or fall) in the future. For that information, students should consult with their academic advisor or see the university course catalog .
- Core Courses
- Thesis Option
Required Core
PHSC 5100 Concepts in Pharmaceutical Science PHSC 5102 Concepts in Pharmaceutical Science 2 PHSC 5212 Research Skills and Ethics or PHSC 6213 Ethical Problems in Health Sciences Research
Medicinal Chemistry & Drug Discovery
CHEM 5626 Organic Synthesis 1 CHEM 5628 Principles of Spectroscopy of Organic CompoundsCHEM 5672 Organic Synthesis 2 CHEM 5676 Bio-organic Chemistry PHSC 5400 Principles of Drug Design PHSC 6222 The Chemistry and Biology of Drugs of Abuse PHSC 6224 Behavioral Pharmacology and Drug Discovery PHSC 6290 Biophysical Methods in Drug Discovery
Please choose electives from the following areas:
Pharmaceutical Sciences (PHSC)
Pharmacology (PMCL)
Pharmaceutics (PMST)
Biology (BIOL)
Chemistry (CHEM)
Nanomedicine (NNMD)
Biotechnology (BIOT)
Thesis credits may count toward the required elective hours.
PHSC 6990 Thesis — this course may be taken twice if necessary
The following course may be taken if additional time is needed to complete the thesis:
PHSC 6996 Thesis Continuation
Admissions Requirements
Prerequisites.
Applicants must have at least two semesters of undergraduate courses (or their equivalent) in each of the following:
- Mathematics (including calculus)
- Biochemistry
- Organic chemistry
Please send all required documents directly to the PharmGrad Application portal . Here is additional information on how to submit documents .
Note: Applicants with deficiencies in their baccalaureate courses may be admitted to the MS Program and allowed to take undergraduate courses at Northeastern University concurrently with certain graduate courses. Students admitted with deficiencies must remove them within the first year of graduate study.
Admissions Checklist
A baccalaureate degree or its equivalent in biology, chemistry, medical technology, pharmacy, chemical engineering or a related field.
A minimum grade point average of 3.0 or higher
Two letters of recommendation (academic and professional)
Personal statement of goals and expectations. Please see application for details.
TOEFL (International Students)
GRE scores are optional
Official transcript from baccalaureate program and all college coursework. Applicants who have degree coursework from institutions outside of the United States must submit a credential evaluation. We require the iCAP WES package ( World Education Services, Inc. ) that evaluates your transcripts course by course.
Got questions?
If you have any additional questions about the graduate program please contact:
Department of Pharmaceutical Sciences

Connect with us
Have more questions about Bouvé? We’re here to help.
Want to take the next step and start your journey at Bouvé?
Request more information
Interested in learning more about what Bouvé has to offer?
Give to the School
UNC Eshelman School of Pharmacy is the No. 1 Pharmacy School in the Nation

The rankings are based on a survey of peers from accredited pharmacy schools across the country and are published every four years.
“I am so proud to be part of a School that relentlessly pursues excellence in education, research, practice and innovation,” said Angela Kashuba, Dean of the UNC Eshelman School of Pharmacy. “This accomplishment is a testament to the unwavering dedication of our students, faculty, staff, alums and partners – they are incredibly committed to our vision and mission.”
“The UNC Eshleman School of Pharmacy’s ranking as the top pharmacy program in the nation comes as no surprise,” said Interim Chancellor Lee H. Roberts. “The School has continually demonstrated excellence by training expert pharmacists, contributing to life-changing research and expanding our health care workforce. Congratulations — this recognition is well deserved once again, and the Carolina community is appreciative of the School’s commitment to our mission of teaching, research and public service.”
Since the last U.S. News & World Report ranking in 2020, the School has adopted a “ Never Still, Never Stopping ” mindset, with a few notable accomplishments:
- Increasing Pharm.D. applications by 56% since 2019.
- Launching the $50 million Research Triangle Center of Excellence in Regulatory Science and Innovation ( Triangle CERSI ), the newest and largest of five CERSI consortiums in the U.S.
- Growing our research enterprise to almost $87 million in research funding for FY23.
- Launching the Master of Professional Science (MPS) in Regulatory Science and new undergraduate minor in pharmaceutical sciences .
- Maintaining the No. 1 residency match rate in the U.S. among pharmacy schools with 75 or more students registering to match.
- Celebrating 10 years of Eshelman Innovation and the launch of a new health tech platform Goldie, which allows for a coordinated post-opioid overdose response for patients in need of care.
- Improving and expanding efforts to inspire, recruit and train the future health care workforce in rural regions of North Carolina through the Rural Pharmacy Health Initiative and rural pharmacy hubs .
For more information about the rankings, visit U.S. News & World Report .
Latest News

Roy Zwahlen receives the 2024 Robert E. Bryan Public Service Award
READ ARTICLE >

Juliane Nguyen inducted into the prestigious AIMBE College of Fellows

Kim Brouwer receives 2024 ASCPT Rawls-Palmer Progress in Medicine Award
Advertisement
Reflections on a 40-year career in drug design and discovery
- Featured Biography
- Published: 03 July 2023
- Volume 32 , pages 1208–1230, ( 2023 )
Cite this article
- Nicholas A. Meanwell 1
4186 Accesses
11 Altmetric
Explore all metrics
In this article, I reflect on a 40-year career in drug discovery at Bristol Myers Squibb that encompassed the cardiovascular, central nervous system and antiviral therapeutic areas. I share scientific observations made in the design and optimization of cAMP phosphodiesterase inhibitors, prostacyclin partial agonists, maxi-K ion channel openers, influenza and respiratory syncytial virus inhibitors, hepatitis C virus NS3/4A proteas, NS5A replication complex and NS5B polymerase inhibitors and inhibitors of HIV-1 attachment and maturation.

Zhongyu Wang, Brain Venables, B. Naidu Narasimhulu, Kevin Gillman, Eric Mull, Timothy Connolly, Louis Chupak, John Kadow, Kyle Eastman, Scott Martin, Jacob Swidorski, Manoj Patel, X. Alan Wang, Paul Scola, Kevin Peese, Omar Lopez, Jeff Romine, Kyle Parcella, John Bender, Rich Hartz, Michael Bowsher, Gan Wang, Xiaofan Zheng, Alicia Regueiro-Ren, Yong Tu, Zhiwei Yin, Zhong Yang, Van Nguyen, Sing-Yuen Sit, Eric Gillis, Makonen Belema, Li-Qiang Sun, Nick Meanwell, Piyasena Hewawasam, Catherine Hathaway, Ny Sin, Yan Chen, Kathy Grant-Young, Ning-Ning Xu, Min Ding, Qian Zhao, Barbara Zheng, Zheng Liu, Tao Wang. Missing are Zhongxing Tim Zhang and Jie Chen.
The virology chemistry team at Bristol Myers Squibb October 19th, 2015.
Avoid common mistakes on your manuscript.
Introduction
My family had no background in studying chemistry or, more specifically, organic chemistry, but I developed an affinity for this branch of science during high school where I focused my last 2 years of study on passing qualifying exams that would enable me to enter university. I was the first in my extended family to attend university and was followed 5 years later by my youngest brother, Neil J. Meanwell, who completed his Ph.D. in inorganic chemistry and who is currently a Professor of Chemistry at Camosun College, Vancouver Island, Canada. Neil’s eldest son, Michael W. Meanwell, has recently started his independent career as an organic chemist at the University of Alberta where he is the Manley and Marian Johnston Professor in Chemistry. I completed my Ph.D. studies at the University of Sheffield in 1979 where I developed synthetic approaches to prostanoids and jasmone under the supervision of Dr. D. Neville Jones, a creative chemist who was also an inspiring mentor [ 1 ]. In early October of 1979, I began a post-doctoral fellowship at Wayne State University in Detroit, Michigan in the laboratories of Professor Carl R. Johnson where I developed aspects of sulfoximine chemistry, including the first preparation and reactions of sulfonimidoyl fluorides, and synthetic approaches to natural products like β-panasinsene and hop ether that, in part, sought to take advantage of sulfoximine chemistry [ 2 , 3 ]. The Johnson laboratory was vibrant with a cadre of bright and capable graduate students that included Jim Zeller, who went on to direct the process group at Parke-Davis in Holland, Michigan, Mike Barbachyn, who would go on to discover linezolid, Tom Penning, who went on to be the lead inventor of celecoxib, John Kadow who was the lead chemist in the discovery of fostemsavir and beclabuvir, and Bob Elliott, who founded J-KEM Scientific. The experiences in these two laboratory settings were both complementary and rewarding, with the challenges encountered providing me with an important foundation for a career in drug design and discovery. I had been attracted to the pharmaceutical industry as a potential career after the compounds that I had prepared during my Ph.D. work were screened by Allen and Hanburys, the respiratory arm of Glaxo Laboratories. However, while this had piqued my interest in pursuing a career in drug discovery, I had only a very limited understanding of the principles of bioactive compound design. Nevertheless, I was able to fulfill that ambition in August of 1982 when I reported to work at the Mead Johnson site of Bristol Myers in Evansville, Indiana where I was to be part of the cardiovascular (CV) drug discovery group reporting to Dr. John E. Lawson. As a drug discovery venue, the Evansville site had originated in 1955 as the “Mead Johnson Research Center” established by the then newly-appointed corporate president D. Mead Johnson. Despite the small size of the enterprise, this site had developed a significant reputation in drug discovery and development by producing several drugs and drug candidates, including the β-adrenergic blocking agents sotalol and bucindolol, the antiarrhythmic agent encainide and the azapirone anxiolytic buspirone. An early and notable contribution to drug design emerged as a consequence of studies by Aubrey (Ole) Larsen who devised the methanesulfonamide moiety as a phenol bioisostere that is a hallmark of the structure of sotalol. My arrival in Evansville coincided with the harmonization of research at Bristol Myers into the “Pharmaceutical Research and Development Division” under the leadership of Dr. Giulio Vita and the retirement of Larsen.
The first project that I was assigned to was to explore analogues of anagrelide ( 1 ), an inhibitor of blood platelet aggregation (BPA) discovered by phenotypic screening that had been advanced into clinical study where it was found to be far more potent in a Phase 1 trial than had been anticipated; unfortunately, the compound caused thrombocytopenia on repeat dosing that extended beyond 4 days and which led to a redirection of the clinical program toward the treatment of polycythemia vera and related orphan diseases [ 4 ]. However, my initial encounter with 1 ended abruptly when Dr. Vita declared a distinct lack of interest in the molecule at the annual program review meeting held 2 weeks after I had started work, a rather blunt introduction to pharmaceutical management decision making that, to a neophyte, seemed to reflect a somewhat mercurial nature that I had not anticipated. I was promptly reassigned before I had even been able to run my first chemical reaction and over the course of the next 12 months, I cycled through three programs in rapid succession, providing for a very frustrating first year, although one that was not without its useful lessons. Two pieces of sage advice that my colleague the late Graham S. Poindexter, an excellent mentor to all who knew him, shared with me during that time was to always conduct reactions on a reasonable scale such that even in the face of a poor yield, there would be sufficient material to complete several rounds of biological screening, of importance in an era where much testing was conducted in animal models of disease. That did not present a problem since that was my natural tendency; however, the second piece of advice, which was to always work with heterocycles, was a little more challenging given that up to that point in my career, my experience of heterocycles had largely been limited to THF and pyridine. Nevertheless, I embraced the philosophy and began to develop knowledge of heterocycle synthesis, properties and function, all of which I came to appreciate far more deeply with the fullness of time and experience.
Michael J. Antonaccio had been recruited to lead the CV discovery group in early 1983 and in the summer of that year, he hired J.J. Kim Wright as head of CV chemistry to whom I was destined to report directly and indirectly for almost the next 20 years. They resurrected interest in 1 and late in the summer of 1983, I was asked to investigate the potential to identify molecules based on an alternate heterocyclic scaffold that would preserve the anti-platelet effects of the compound, which by then had been determined to be a function of inhibition of platelet cyclic adenosine monophosphate (cAMP) phosphodiesterase (PDE), but divorce from the thrombocytopenia. The latter was a seemingly formidable challenge since there was no preclinical model of the side effect, despite a survey of more than a dozen species, and no predictive in vitro assay; consequently, the approach pursued was one of identifying a novel chemotype that would be tested in humans based on the belief that an alternate heterocycle might mitigate the toxicity. Working initially as a lone chemist and under the threat of being moved to yet another project if I did not produce a lead within 6 months, we targeted a series of molecules that were to be prepared as ungarnished prototypes. These would then be equated with the modest but detectable potency of the parent 1,2,3,5-tetrahydroimidazo[2,1- b ]quinazolin-2-one (i.e. 1 lacking the two chlorine substituents), a useful strategy that I continue to consider an effective approach and advocate for where appropriate. The parent 1,3-dihydro-2 H -imidazo[4,5- b ]quinolin-2-one 2 , a molecule with low prevalence in the literature that was restricted to just two published articles, both of which were >30 years old and which merely described synthetic approaches, emerged as the lead chemotype. The initial sample of 2 exhibited poor solubility properties, not unlike the tetrahydroimidazo[2,1- b ]quinazolin-2-one series, and frustrated with trying to isolate pure material from the partially saturated 9,9a-dihydro precursor, I handed J. Stuart Fleming a sample of the mixture and asked him to test the compound prior to submitting it through the formal channels. He called me a few days later to inform me that the compound was an inhibitor of ADP- and collagen induced rabbit BPA with potency comparable to the anagrelide prototype [ 5 ]. Not surprisingly, this provided an impetus to rapidly develop a process to access the fully oxidized compound which reproduced the platelet inhibitory profile. We embarked on a broad synthesis campaign with assistance from a team that began to grow as the program evolved and BMY-20844 ( 6 ), the 7,8-dimethyl derivative, was identified as a compound suitable for advancement into clinical study. However, synthetic access to these molecules was long and laborious until we devised the Wadsworth-Emmons reagent 4 , a compound obtained straightforwardly by bromination of hydantoin followed by an Arbuzov reaction with triethyl phosphite, a process that was exothermic in nature. The phosphonate 4 proved to be a highly effective, reliable and practically convenient reagent for the preparation of C-5 substituted hydantoin derivatives, as depicted in Scheme 1 [ 6 ]. Phosphonate 4 would be my first marketed product since it was commercialized by Lancaster Synthesis after we had published the reactivity profile of the compound. The reaction of 4 with the activated but sterically encumbered aldehyde 3 provided 5 in 86% yield as a single isomer via a procedure that was both rapid and preparatively straightforward, with the product precipitating in essentially pure form after diluting the reaction mixture with water. The identity of the product was confirmed by measuring the long range 1 H- 13 C coupling constant between the vinyl proton and the C-4 carbonyl carbon atom in the fully coupled 13 C nuclear magnetic resonance (NMR) spectrum with the J value larger for the trans relationship than the cis [ 6 ]. The subsequent hydrogenation of 5 , cyclization of the aniline onto the hydantoin C-4 carbonyl and oxidation to afford 6 was uneventful, benefiting from all of the optimization work that we had done up to that point. This preparative procedure was adopted by the process group to make the material needed for investigational new drug (IND) toxicological and clinical studies [ 5 ]. The development of this synthetic methodology advanced the program significantly, providing rapid, convenient and reliable access to a wide variety of analogues with differing functionalities whilst also offering an important lesson in program prosecution. Another important lesson that I learned in this year of study was that the identification of a proprietary chemotype provided for the ready adoption of emerging discoveries from others who had begun to explore the anagrelide chemotype, with several patent applications appearing in the second year of our effort. The advantage offered by the introduction of the amide-containing side chain observed with lixazinone ( 7 ) transferred to the imidazo[4,5- b ]quinolin-2-one chemotype 2 , with 8 offering enhanced potency while 9 provided a homologue with enhanced aqueous solubility, an enduring challenge for both series [ 7 , 8 ]. This highlighted the value of a proprietary chemotype to allow for the rapid interpretation and implementation of competitive developments and was a compelling strategic insight that has endured. Unfortunately, in a Phase 1 dose escalation clinical study, 6 failed to impart a significant effect on ex vivo BPA inhibition, presumably a function of absorption and pharmacokinetic (PK) issues that were not fully elucidated. It is also not clear that 6 would have resolved the thrombocytopenia side effect associated with 1 , the mechanism of which is only now beginning to come into focus 50 years after the compound’s discovery [ 4 ]. Anagrelide ( 1 ) interferes with megakaryocyte differentiation and proplatelet formation and has been shown to stabilize a complex between a dimer of cAMP PDE3A and a dimer of the RNase Schlafen 12 (SLFN12), acting as a molecular glue that enhances the half-life of SLFN12 in cells whilst favoring the active, dephosphorylated form of the enzyme. The role and function of the Schlafen proteins, of which there are six expressed in humans, in physiology and disease is still emerging and precisely how this observation relates to the thrombocytopenic effects of 1 remains to be determined. Interestingly, the recent developments that uncovered this biochemical pharmacological effect was the result of phenotypic screening, providing a compelling example of this approach to drug discovery whilst reflecting an interesting symmetry given that 1 was also discovered by a phenotypic screen [ 4 ].

Synthesis of BMY-20844 ( 6 )
As the chemistry lead for what I viewed to be a developing anti-thrombotic franchise and with a drive to be successful in providing drug candidates, I engaged in developing a deeper understanding of the mechanisms of platelet function and the broader field of thrombosis whilst enhancing my awareness of competitive activity. Consequently, I began to prepare select compounds appearing in the literature that were claimed to be broad-spectrum inhibitors of BPA, the attribute that distinguished 1 and the imidazo[4,5- b ]quinolin-2-ones 6 , 8 and 9 from the majority of the most prominent anti-platelet agents at the time, which were generally active against a narrower range of stimuli. The value of this approach was further enhanced by its potential to identify an alternate chemotype or mechanism of action that would fulfill our target profile should the imidazo[4,5- b ]quinolin-2-one series fail to deliver on objectives, a concern that I subsequently learned would be a constant companion. This was another important and enduring insight that I developed and one that would subtend strategic decisions in many subsequent projects where either an alternate scaffold or mechanism of action (MOA) offered the potential for the rapid relief in the face of an unanticipated challenge. While almost all of the literature compounds that I prepared failed to meet expectations, one that did was the acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor octimibate ( 10 ), which I prepared in the summer of 1987 [ 9 ]. Octimibate ( 10 ) exhibited potency comparable to the parent imidazo[4,5- b ]quinolin-2-one 2 and, interestingly, human platelets were 10-fold more sensitive to inhibition induced by ADP than rabbit platelets [ 10 ]. Preliminary studies by my biochemical pharmacology collaborator, the late Steve Seiler, indicated that 10 was enhancing cAMP levels in platelets, providing an explanation for the broad spectrum of inhibitory action [ 10 ]. Despite the absence of a defined mechanism of action and with the tacit approval of Kim Wright, I began to develop a plan to explore 10 in more detail. In a search for inspiration for design concepts, I sat down one Saturday afternoon in early October of 1987 and perused every molecule in the United States Adopted Names (USAN) dictionary seeking ideas for structural motifs that had some overlap with 10 and which might be gainfully explored. Whilst this exercise provided me with an excellent opportunity to review the existing pharmacopeia and assimilate the general complexion of drug molecules, a more important facet for me was that I would be reviewing and selecting chemotypes and functionalities from a body of molecules that had some form of validation as a drug or drug candidate. In effect, I was unwittingly following the wisdom and advice of Sir James Black who said “The most fruitful basis for the discovery of a new drug is to start with an old one”, a comment that I encountered only much later, although I have been unable to precisely pinpoint its origin [ 11 ]. One molecule that I found intriguing and appealing based on its relative simplicity was the non-steroidal anti-inflammatory drug (NSAID) oxaprozin ( 11 ) which incorporated a 4,5-diphenyloxazole ring, a heterocycle that I had not previously encountered, and a propionic acid side chain. After perusing the literature associated with oxazoles, which was more of a burden then in the absence of digital literature searching, and learning how they were constructed, I began the campaign by synthesizing the nonanoic acid homologue of 11 which inhibited human BPA with EC 50 values of 1.4–2.5 µg/ml, about 10-fold less potently than 10 [ 12 ]. With the view that we may need an azole heterocycle with 4 vectors that would more effectively mimic those of 10 , we adopted pyrazole as a workhorse heterocyclic chemotype with which to explore structure-activity relationships (SARs), a decision that was instrumental in deducing fundamental aspects of the molecular topology of the pharmacophore [ 13 ]. A few months into the campaign, Steve Seiler called me one morning to inform me that he and his team had elucidated that 10 was functioning as a partial agonist at the prostacyclin (PGI 2 ) receptor, which explained the observation of elevated cAMP in platelets while enhancing the appeal of the chemotype since it would act synergistically with cAMP PDE inhibitors [ 10 ]. Interestingly, I had not seen Steve face-to-face for about a week, an unusual occurrence that, in this case, had coincided with me experiencing a reaction to being exposed to some of these compounds. I had experienced a form of facial flushing and swelling, with a reddening of the skin but with no discomfort and no other symptoms that might be attributed to PGI 2 agonism. When Steve called me, the effect had begun to wane and my skin had taken on a sort of waxy pallor whilst my new associate, Michael Rosenfeld, was beginning to experience some skin sensitivity with the appearance of reddened blotches. The synthetic accessibility of these early compounds was such that, at this point in the effort, we were typically preparing five or more final compounds a week on gram scale and the PGI 2 agonism provided a potential explanation for the symptoms. While the effect may have been related to the specific compounds being made at that time, we exercised greater caution and a heightened awareness of the properties of the compounds more broadly, with the result that, for us, there was no significant reoccurrence. Although we explored a wide range of azoles, an exercise that provided us with practical insight into their synthesis, attributes and physicochemical properties, we adopted the 4,5-diphenyloxazole as the main vehicle for drug design based on its properties and synthetic accessibility. By installing aromatic rings in the side chain of the nonanoic acid lead, we hoped to constrain conformational mobility as a means of pre-organizing the molecule for optimal receptor recognition, a strategy that ultimately led to the design of BMY-45778 ( 12 ) as the most potent and defining compound of the series, which was prepared by Jeff Romine [ 14 , 15 ]. Interestingly, the single crystal X-ray structure of 12 revealed that the phenoxy ring, both oxazole heterocycles and one of the phenyl rings attached to the terminal oxazole adopted a planar topography, with just a 10 o variation from planarity (Fig. 1 ), a topography also found to be present in solution based on 1 H-NMR analyses that were enabled once we had a developed the necessary cadre of molecules to study [ 14 ]. This was an important element in molecular recognition since analogues not able to access a planar topographical arrangement were much less potent, indicative that the binding pockets of some targets demand planarity in their ligands. This is an interesting insight in an era when escaping from the flatlands has been persistently advocated and to which I am fully sympathetic [ 16 ]. Interestingly and, somewhat surprisingly given the partial agonist nature of these compounds, we were not able to identify a PGI 2 antagonist from the large and diverse collection of molecules that we had synthesized throughout the program.

Single crystal X-ray structure of 12 depicting the planar topography (Cambridge Structural Database (CSD): PIDWII, 1233161)
My anti-thrombotic era came to an end in late 1991 following the final consolidation of research under the new Bristol Myers Squibb (BMS) organization where the priorities of the cardiovascular group were directed toward thromboxane receptor antagonists, as exemplified by ifetroban ( 13 ). The 1,3-dihydro-2 H -imidazo[4,5- b ]quinolin-2-one program ended with a failure to identify compounds with targeted metabolic stability while the non-prostanoid PGI 2 agonist project ended with studies conducted in cynomolgus monkeys who were found to be poorly tolerant of orally administered 12 . However, the non-prostanoid PGI 2 agonist chemotype was of interest to other companies and the pharmacophore that we had painstakingly mapped out is represented in selexipag ( 14 ), a prodrug discovered by Nippon Shinyaku and developed clinically by Actelion for the treatment of pulmonary arterial hypertension (PAH) [ 17 ]. This compound was part of the $30 billion dollar acquisition of Actelion by Johnson and Johnson in 2017 and is currently marketed as Uptravi®, with the value of the drug for those suffering from PAH reflected in worldwide sales that reached $1.322 billion in 2022. In addition, United Therapeutics is currently evaluating ralinepag ( 15 ) in Phase 3 clinical trials, also for a PAH indication [ 18 ].
With the consolidation of research at the new Bristol Myers Squibb organization, which meant that the CV group would be based in Lawrenceville, New Jersey, I availed myself of the opportunity to move into the central nervous system (CNS) arena where I hoped to expand my therapeutic area experience and further advance my medicinal chemistry knowledge and skills. There I began collaborating with Val Gribkoff, a scientifically rigorous electrophysiologist who was advocating for studying openers of the large conductance, Ca 2+ -dependent potassium channel known as maxi-K as an approach to enhancing cellular hyperpolarization that would protect neurons against the excitotoxic cell death that follows a stroke and contributes to the development of the ischemic penumbra [ 19 ]. The benizimidazol-2-one derivative NS-004 ( 16 ) was a prototype maxi-K opener that was adopted as a lead molecule after confirming its activity in electrophysiology assays and we began to develop a plan to explore and understand the channel opening pharmacophore inherent to the compound. In contemplating 16 as a design template and in the complete absence of any SAR information, we initially assumed that the secondary amide, the phenol moiety and the CF 3 substituent were important contributory elements to the pharmacophore and these were preserved in the concepts that we developed, which were designed to provide additional opportunities for structural elaboration. The initial design concepts focused on exploring the relationship between the phenolic hydroxy substituent and the heterocyclic core which, by virtue of the single rotatable bond in the molecule, suggested the potential for either an orthogonal arrangement, represented by 3-hydroxy oxindoles 17 , or a coplanar arrangement where we designed templates that projected the phenol and C=O moieties in proximity, as represented by 18 and 19 . In addition, we contemplated the two topologically complementary deannelative arrangements captured in 20 and 21 and the effect of altering the functional group relationships in the stretched analogue 22 . These concepts provided ample opportunity to explore the substitution pattern of the phenol and heterocyclic rings while success with one of the concepts would allow introduction into the others, although with some interpretation dependent on the structural background. However, the ability to explore many of these concepts required the development of synthetic methodology to access key building blocks that, at that time, were not available commercially. A particularly important intermediate was 6-trifluoromethyl isatin ( 25 ), a precursor to 17 – 19 that could not be accessed by the classic Sandmeyer synthesis because formation of the heterocycle ring element relied upon a ring closure reaction that was dependent on the nucleophilicity of the aromatic ring. This problem was solved by combining ortho -directed metalation of tert -butyl (3-(trifluoromethyl)phenyl)carbamate ( 23 ) to generate the lithium anion which was reacted with diethyl oxalate to afford 24 which, in turn, was cyclized to give 25 under aqueous acidic but not anhydrous acidic conditions, as summarized in Scheme 2 [ 20 , 21 , 22 ]. A new synthetic approach to the selective functionalization of benzimidazole-2-one that provided access to analogues of 22 was also developed as part of this campaign [ 23 ].

Synthetic approach developed to access 6-trifluoromethyl isatin ( 25 )
In the first year of the maxi-K program, compound screening proved to be laborious because of a reliance on testing compound effects on native maxi-K channels in patches isolated from neuronal cell membranes where these channels were not sufficiently abundant to allow routine evaluation. Although a possible source of frustration, we took advantage of this opportunity to explore all of the design concepts that had been conceived which we were able to complete in relatively short order. With the obtention of a clone of the maxi-K channel RNA from mouse ( mSlo ) and, subsequently, human ( hSlo ) that expressed the channel proteins in Xenopus oocytes , we found ourselves in a position to screen prototypical representatives of each chemotype and select the optimal leads for further study. There was thus an absence of the pressure to pursue the first positive lead molecule that came along, a circumstance that allowed for the removal of the kind of bias that had the potential to shape the overall trajectory of the program. We were able to comprehensively survey the various chemotypes with, remarkably, channel-opening activity observed in almost all of them. While the stretched chemotype 22 provided active maxi-K openers and delineated fundamental SARs which revealed a requirement for an electron withdrawing substituent on the phenyl ring of the heterocycle, the oxindole chemotype 17 offered improved efficacy at higher concentrations and was pursued more vigorously [ 24 , 25 , 26 , 27 ]. The 3-hydroxy-oxindole derivatives were active and also required an electron withdrawing substituent on the heterocycle for expression of channel opening activity, while the 3-fluoro homologues demonstrated improved CNS penetration. Flindokalner ( 26 ), devised and synthesized by Piyasena Hewawasam, was ultimately selected for clinical study and subsequently advanced into a Phase 3 clinical trial for evaluation as an intravenously (IV)-administered treatment for the improvement of symptoms following a stroke [ 19 , 25 , 26 , 27 ]. However, it was shortly after the discovery of 26 that I was recruited by Kim Wright to lead virology chemistry and John Starrett assumed responsibility for the maxi-K chemistry team, collaborating with Val Gribkoff to complete the preclinical profiling of 26 . John subsequently addressed the limited aqueous solubility of 26 with the phosphonooxymethyl prodrug 27 and identifying the 1,3,4-oxadiazol-2(3 H )-one 28 and its prodrug 29 as back-up compounds [ 28 , 29 , 30 ]. The discovery working group also advanced the quinoline-2-one 30 into clinical study for the treatment of erectile dysfunction and the triazolone 31 for the treatment of urinary incontinence [ 31 , 32 ]. Unfortunately, 26 failed to demonstrate a significant activity in ameliorating the effects of an ischemic stroke, adding yet another mechanism to the broad palette of failure for this devastating disease, whilst 30 and 31 ultimately failed to advance to late-stage clinical studies [ 33 ].
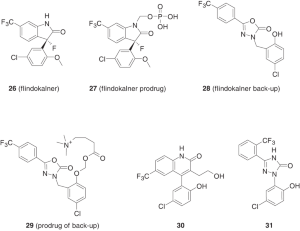
One interesting opportunity that emerged from this program was when Val Gribkoff examined the effects of 16 on the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-dependent small conductance chloride channel that had first been cloned in 1989 [ 34 ]. In Xenopus oocytes expressing wild-type (WT) CFTR, concentrations of 16 ranging from 10–30 µM rapidly and reversibly enhanced current to 30–60% of the maximal effect of cAMP. The effect of 16 was muted when tested against the ΔF508 mutant channel with <20% increase in current compared to WT but co-application with cAMP produced an enhanced effect. In a collaborative effort conducted in Michel Lazdunski’s laboratory near Nice, France, the effects of 16 on WT and ΔF508 CFTR were reproduced and extended to Vero cells expressing the chloride channels, with the data suggesting that the drug targeted the phosphorylated form of the channel [ 34 ]. Although this study presented an interesting opportunity for drug discovery that would ultimately be addressed more than a decade later with the development of clinically effective channel potentiators (channel openers) and channel trafficking correctors, after careful deliberation the company chose not to pursue therapeutics directed toward cystic fibrosis, a disease management area that fell outside of the existing portfolio and focus [ 35 ].
The structural diversity of the clinical compounds to emerge from the maxi-K program reflects the benefit of developing the broad range of scaffolds that were explored in the first year of the campaign and provided an excellent foundation for the considerable success the team enjoyed in identifying clinical candidates that targeted both the CNS and periphery. In contrast, my life in virology during this period was considerably bleaker as we sought to continue to develop mechanistically novel human immunodeficiency virus-1 (HIV-1) inhibitors and to build a preclinical and clinical franchise around inhibitors of both hepatitis C virus (HCV), which had just been characterized molecularly, and respiratory viruses. Using a phenotypic screening approach, Mark Krystal and his team had discovered and characterized the influenza virus fusion inhibitor 32 and Milind Deshpande, who was leading the burgeoning library synthesis group, devised and constructed a small library of 175 compounds in which the amine moiety was varied. This initiative identified the cyclohexanol 33 as a compound with enhanced antiviral potency compared to the prototype, with the axial isomer 34 4-fold weaker. This result provided an excellent lesson in bias in drug design since I had assumed that the amine in 32 was likely to be pharmacophoric. However, the library prepared by Milind and his team demonstrated the potential of focused combinatorial chemistry to rapidly explore a pharmacophore in an unbiased fashion. The amine discovered with 33 and 34 was then used to interrogate potential salicylic acid replacements which identified a thioamide derivative that, interestingly, was the result of reaction at an alternative site of the substrate to that anticipated. In this chemotype, the axially-disposed isomer 36 was considerably more potent than the equatorial isomer 35 , the reverse of the SAR associated with 33 and 34 [ 36 , 37 , 38 , 39 ]. Unfortunately, the lead molecule 32 exhibited antiviral activity toward only the H1 and H2 hemagglutinin (HA) subtypes and despite an extensive study around the chemotype that was expansive in nature, we were never able to achieve significant inhibition of the H3 virus subtype. A detailed scientific examination that was able to combine resistance mutation data with labeling by an azide-based photoaffinity probe that generated a reactive nitrene intermediate on irradiation, provided an understanding of the observed profile. The photoaffinity labeling experiments presented a challenge but the broad-based SAR survey that had been conducted along with experiments that had identified inhibitors of the HA inhibitors indicated that under the acidic conditions of the assay, the compounds were trapped in the HA protein and could not readily be displaced. This suggested conducting the photolabelling experiments at low pH, an experimental design that more effectively captured the active site residues with the nitrene probe. This allowed us to propose that the salicylamide moiety functioned as a carboxylic acid bioisostere by engaging with the side chain guanidine of an arginine residue that was present in H1 and H2 HA but not in H3 HA, where the residue was a histidine, much less basic and lacking the two H-bond donors that would complement 32 – 34 . With the inability to identify a viable chemotype with broad spectrum influenza inhibition, we abandoned this program after 18 months. However, while were unsuccessful in identifying broad spectrum influenza inhibitors, the scientific discoveries and insights that were gleaned were interesting and proved to be informative for later programs, particularly in HCV.
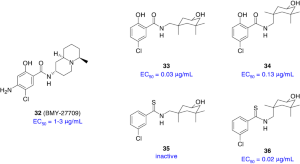
The virology biology and chemistry groups spent the next few years devising and implementing a range of screening assays for several viruses of interest and exploring medicinal chemistry approaches to biochemical targets. However, we had little to show for our effort although under Rich Colonno’s leadership of virology, entecavir ( 37 ) was discovered to be a potent hepatitis B virus (HBV) inhibitor and the HIV-1 protease inhibitor atazanavir ( 38 ) was licensed in, both of which went on to be highly successful drugs [ 40 , 41 ]. Persistence is an important trait, although it can be abused and, despite the limited library of compounds available for screening at that time, our proclivity toward phenotypic screening ultimately proved to be fruitful. Mark Krystal and his team discovery the respiratory syncytial virus (RSV) fusion inhibitors 39 and 40 while a screen devised by Pin-fang Lin and Wade Blair identified the prototype HIV-1 attachment inhibitor 41 , both of which acted on viral coat proteins, although with quite different and novel MOAs [ 42 , 43 , 44 , 45 ]. The RSV inhibitors 39 and 40 had been in the compound collection for over 30 years and had experienced many screening campaigns without producing a positive effect, thus largely being viewed as dark matter prior to our discovery. The glyoxamide 41 was a member of a commercial library of amides and sulfonamides that had developed some notoriety within BMS for its low fidelity of purity, with several samples still containing acid chloride substrates that would often be active in a screen but of no redeeming value as lead molecules. Fortunately, we were able to reproduce the inhibitory activity of 41 with freshly synthesized material and this molecule proved to be a bona fide lead that we would spend more than 6 years investigating and developing.
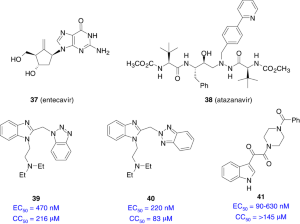
Careful profiling of these leads confirmed their validity and both spawned full phase optimization campaigns, with the RSV inhibitor program ultimately identifying BMS-433771 ( 42 ) as a clinical candidate although not after some travail [ 46 ]. To demonstrate proof-of-concept for antiviral effect in the cotton rat model of infection, we had to resort to a topical delivery mode in which the animals were exposed to an aerosol of the drug for 22 h a day. This required 20 gram quantities of inhibitor with a water solubilizing element, which the pharmacophore fortunately tolerated. Kuo-Long Yu and the team provided these materials with seemingly apparent ease, with synthetic accessibility to the benzimidazol-2-one derivatives that had become the workhorse chemotype benefitting from the earlier methodology development [ 23 ]. MOA studies with this chemotype relied upon the characterization of viruses grown to be resistant to 42 and were complemented by the development of a diazirine-based a photoaffinity probe that was designed to generate a reactive carbene to label the virus target protein. These studies initially identified the fusion (F) protein as the target, while labeling studies conducted with elements of the purified F protein suggested that the molecule interfered with 6-helix bundle formation, a critical step that occurs during the later stages of the membrane fusion process [ 47 ]. Interestingly, subsequent crystallographic studies conducted by others pursuing RSV fusion inhibitors based on this chemotype and others suggested that they bound to the core of the intact fusion protein trimer, stabilizing it toward the conformational changes essential for triggering membrane fusion and a productive infection [ 48 ]. Unfortunately, a change in strategic direction at BMS led to 42 being abandoned just 1 month before the IND application was anticipated to be filed. While this was a disappointment for a team that had overcome several significant challenges, the chemotype and MOA attracted the attention of others and both riletamovir ( 43 ) and sisunatovir ( 44 ) have demonstrated clinical efficacy in experimental infections in normal healthy volunteers (NHVs) [ 48 , 49 , 50 , 51 , 52 ]. While 43 was advanced into Phase 3 clinical studies, it appears to have been abandoned by its sponsor in 2022. The FDA granted Fast Track designation to 44 in 2020 and in 2022 the compound was acquired by Pfizer as part of a transaction with a value of up to $525 million [ 53 ].
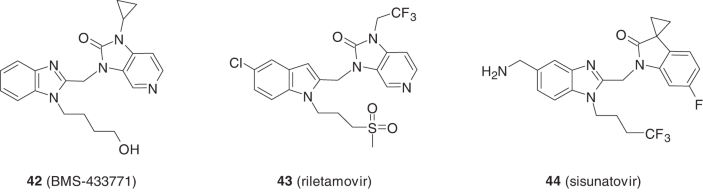
The HIV-1 AI program presented unique challenges associated with the gp120 target, with variability in the sensitivity of a number of viruses despite the good sequence conservation in the putative binding site pocket. HIV-1 gp120 is a protein that exploits conformational mobility as one element in avoiding immune surveillance, with glycation (the glycan shield) and the ability to generate resistance mutations being the other two methods. Surveying the SARs associated with the lead HIV-1 AI 41 using the pseudovirus assay that had identified the hit molecule moved quickly initially based on the relative ease of exploring variations to the benzoyl moiety; however, molecular edits at this site were poorly tolerated and this motif was maintained throughout much of the program. Considerably greater success was achieved by substituting the core indole from which the 4-fluoro and 4-methoxy analogues were 50- and 300-fold more potent than 41 , respectively. The C-5 and C-6 positions of the indole heterocycles were poorly tolerant of substitution but C-7 was not, and the C-4, C-7-dimethoxy indole derivative exhibited an EC 50 of 250 pM in the pseudovirus assay. However, the intrinsically poor aqueous solubility of the chemotype was a persistent challenge that was addressed, in part, by exploring azaindole derivatives that ultimately focused on the 6-aza chemotype since that allowed substitution at both C-4 and C-7 [ 54 ]. Prosecution of the program required the development and/or optimization of a number of synthetic methodologies in order to access the full panoply of target molecules as we wrestled with the challenges presented by potency variation and the inherent pharmaceutical properties [ 55 ]. BMS-488043 ( 45 ) emerged as a clinical candidate that provided proof-of-concept (POC) for the HIV-1 AI mechanism in a Phase 2a clinical study where there was a dose-related reduction in viral load, although concomitant administration of the drug with a high fat meal was required in order to drive plasma exposure, a function of solubility- or dissolution-limited absorption problems with the molecule. While 45 was advancing in the clinic, optimization continued and under John Kadow’s leadership we found that a heterocycle substituent installed at C-7 was optimal, with those azoles and azines that were capable of adopting a coplanar arrangement with the azaindole core the most potent. This finding built upon observations made by Kap-Sun Yeung with the indole chemotype with C-7 carboxamides in which the dimethyl-amide was substantially less active than the mono-methyl or primary amide, interpreted as a need for coplanarity between the core and the substituent. This was another example of pharmacophore favoring planarity and since N -linked azoles offered an advantage in PK profiling assays, temsavir ( 46 ) was selected as a clinical candidate and advanced into Phase 1 study. However, the need for a high fat meal to drive the plasma exposure of 45 precipitated a campaign to identify a solubility-enhancing prodrug, with a phosphonooxymethyl moiety fulfilling the needs of the program, demonstrating excellent dose escalation in preclinical studies and abrogating the food effect. Phosphonooxymethyl prodrugs of both 45 and 46 were prepared as well as an additional analogue of the latter compound, with salt selection important for compound isolation and chemical stability. The application of this kind of prodrug is best suited to BCS class II compounds that exhibit high membrane permeability but low aqueous solubility and requires a delicate balance of drug properties and release, which occurs presystemically and is mediated by alkaline phosphatase at the brush border membrane in the gastrointestinal (GI) tract, to ensure that drug is absorbed before precipitating in the gut. Fostemsavir ( 47 ) was accepted as a clinical candidate along with two other AI prodrugs, with one falling out in IND toxicology studies when the parent drug precipitated in tissues, a function of its intrinsically low aqueous solubility combined with the ability of the phosphonooxymethyl prodrug technology to very effectively deliver drug to plasma and tissues. The success of this phosphonooxymethyl-based prodrug approach, for which precedent had largely been restricted to IV drugs, led to the formation of a team at the Biocon-Bristol Myers Squibb Research Center in Bangalore, India dedicated to developing expertise in prodrug design and exploration to provide solutions to a broad range of developability challenges across the portfolio. In addition, in order to further enhance the awareness of prodrug technology within BMS, I partnered with my colleague Dinesh (Dolatrai) Vyas from oncology, who had been intimately involved in the discovery of etoposide phosphate and other anticancer prodrugs, to publish an internal quarterly newsletter that captured emerging developments. Collaborating with Dinesh on this initiative was immensely rewarding at many levels since he is an excellent scientist who brought new dimensions to my knowledge and understanding of medicinal chemistry. Moreover, I not only benefited directly from his mentorship on this enterprise, but I was an indirect recipient of his mentoring skills through the several members of my team that he had nurtured through the early phases of their careers.
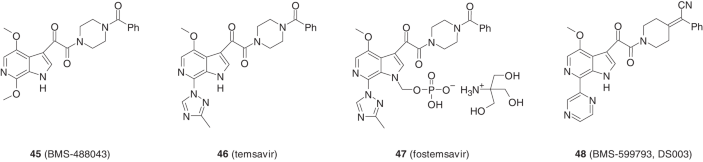
The capacity of 47 to deliver 46 in vivo in preclinical study was largely recapitulated in Phase 1 clinical trials, with the prodrug allowing dose escalation to targeted plasma levels that had been set after evaluation of 46 against viruses available from clinical studies with the protease inhibitor 38 . Targeted plasma levels were set at a concentration that would inhibit 90% of the circulating viral isolates by 90% (EC 90 ). However, the improved delivery of 46 from 47 unmasked PK deficiencies that had not been observed in preclinical studies, and it was clear that 46 exhibited a short plasma half-life in humans. This profile had been masked by flip-flop kinetics when administering 46 directly, a function of slow absorption due to the intrinsically poor aqueous solubility that had prolonged the absorption phase. At this point, Peter Timmins and Jonathan Brown at the BMS site in Moreton, England stepped in to develop an innovative slow-release formulation that relied upon the combination of a specialized site-of-absorption study and a novel in silico modeling approach to build an understanding of the drug release and absorption characteristics required to reliably deliver the drug on a bis-in-die (BID) dosing schedule [ 56 ]. This initiative was successful and provided a path forward for the Phase 3 BRIGHTE clinical study which was completed by ViiV Healthcare who had acquired 47 in 2016 [ 55 , 56 ]. Fostemsavir ( 47 ) was approved by the United States (U.S.) Food and Drug Administration (FDA) on July 2nd, 2020 after being designated as “Breakthrough Therapy” and accorded “Fast Track” and “Priority Review” status to expedite review [ 57 ]. Approval in the European Union followed in February of 2021 and the drug is marketed as Rukobia® for highly treatment-experienced adults with multidrug-resistant HIV-1 who are failing their current antiretroviral regimen due to resistance, intolerance, or safety considerations. Fostemsavir ( 47 ) was the first HIV-1 drug with a new MOA to be introduced in the 15 year period from 2007 to mid-2022 and the interesting clinical profile of this molecule continues to be developed and understood [ 57 , 58 , 59 ]. The HIV-1 AIs have also been shown to be effective microbicides in non-human primates, particularly when used in combination with other virus entry inhibiting drugs, and BMS-599793 (DS003, 48 ) was licensed to the “International Partnership on Microbicides” who are studying the molecule for its potential to act as a topically-applied microbicide to prevent HIV-1 infection [ 60 ].
HCV was an important disease target from my very early days in virology but the absence of cell-based replication assays severely blunted inhibitor identification and characterization. We anticipated from the outset that combination therapy would be required to prevent the emergence of resistance since HCV was comprised of an RNA genome and there already existed considerable virus diversity with, at the time, 6 known genotypes (GTs) and multiple subtypes. GT1a was most prevalent virus in the U.S. and Europe while GT1b dominated in Asia. The development of in vitro biochemical assays had focused on the HCV NS3/4A protease and the NS5B RNA-dependent, RNA-polymerase (RdRp), antiviral targets with appeal based on prior experience with HIV-1 therapeutics, that were eventually enabled by X-ray crystal structure data. These assays were, by definition, approximations of the natural state in cells but were nevertheless useful for identifying lead inhibitors. The HCV NS3/4A protease was almost sloth-like in its processing activity compared to mammalian serine proteases and proved to be a challenging target, with an active site described as featureless or, as Carl Decicco, my supervisor after the DuPont acquisition, liked to say, dimples on a golf ball. We explored saccharin and related activated carbonyl derivatives that were known mechanism-based serine protease inhibitor chemotypes that led to the discovery of the potent and selective inhibitors of mast cell tryptase 49 and 50 but not of HCV NS3/4A protease [ 61 ]. We subsequently engaged in a collaboration with Axys Pharmaceuticals designed to take advantage of their intriguing delta technology in which serine protease inhibitors that bound to the active site presented functionality with a geometry that, in conjunction with the catalytic serine oxygen and histidine nitrogen atoms, created a coordination site for Zn 2+ that would stabilize the complex. However, this identified only intractable inhibitors like 51 while the several series of peptide-based approaches that we explored were unsuccessful [ 62 ].
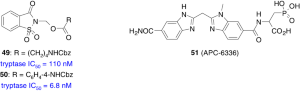
However, our prospects began to change in January of 2000 with the publication of a series of patent applications from Boehringer-Ingelheim that disclosed tripeptide-based inhibitors of HCV NS3/4A protease. These molecules were distinguished by the presence of a large P 2 * substituent attached to the P 2 proline residue while the P 1 moiety was a substituted cyclopropylglycine and the C-terminus was a carboxylic acid moiety. These molecules were the result of a classic and painstaking medicinal chemistry optimization campaign that capitalized on the observation of product inhibition and provided an important foundation for HCV NS3/4A inhibitor design as well as the first clinical candidate based on this chemotype, BILN-2061 ( 52 ) [ 63 ]. This chemotype provided us with a suitable vehicle to explore the concept of exploiting the small but well-defined S 1 ’ pocket of the enzyme, an idea that had been conceived several months earlier as we were winding down the collaboration with Axys Pharmaceuticals.

The concept that had been envisioned was to access the S 1 ’ site by exploiting an acylsulfonamide moiety that would preserve the acidic element that engaged the catalytic residues while providing for the read-through topology to project a substituent into the S 1 ’ pocket. After having broken our pick twice on HCV NS3/4A protease as a biochemical target, we had been reluctant to propose exploring the concept in the context of the hexapeptide derivatives replete with several carboxylic acid moieties that were the most effective inhibitors at the time. We prepared the prototype acid 53 (Table 1 ) from the patent applications and were able to reproduce the claimed potency, with the simplest iteration of the design concept, the methanesulfonamide 54 , fully preserving the inhibitory activity of the progenitor [ 64 ]. However, guided by computer-aided drug design (CADD), the cyclopropyl homologue 56 offered significantly enhanced potency both in the biochemical enzymatic assay and, most particularly, in the GT-1b replicon assay that had just been developed by my colleague Min Gao and his team. X-Ray cocrystal structure data revealed that both of the sulfonamide oxygen atoms were involved in drug target interactions while the cyclopropyl ring optimally filled the S 1 ’ pocket [ 65 ]. The iso -propyl homologue 55 , which results from the addition of just two hydrogen atoms to 56 , was 20-fold less potent in both assays, reflecting the importance of correctly occupying the S 1 ’ pocket, while the cyclobutyl ( 57 ) and cyclopentyl ( 58 ) homologues were progressively weaker inhibitors than 56 .
BMS-605339 ( 59 ) was the first candidate advanced into clinical study and demonstrated dose-dependent efficacy at reducing viral load following oral administration to HCV-infected patients [ 64 ]. However, the compound was associated with mild bradycardia, PR interval prolongation and junctional rhythm disturbance, cardiac side effects that, although asymptomatic in nature, gave rise to significant safety concerns that, after considerable debate, was sufficient to terminate the clinical development of 59 [ 64 , 66 ]. Humans were >50-fold more sensitive than preclinical species to the CV effects of 59 which could be recapitulated in a Langendorff isolated rabbit heart assay where heart rate decline and sinoatrial node recovery time (SNRT) prolongation were monitored, with test compounds assessed at a concentration of 10 µM. Despite the low throughput nature of this assay, Paul Scola and his team were able to identify BMS-650032 ( 60 ) within less than 20 structure-function iterations and concomitant with the decision to terminate 59 , a remarkable example of a combination of careful medicinal chemistry analysis, inspired drug design and intuitive interpretation of structure-function relationships. The structural differences between 59 and 60 are subtle in nature, with just a single chlorine atom added to the molecular formula and an alteration of the substitution pattern of the isoquinoline ring, but profound with respect to the cardiac liability profile. BMS-650032 ( 60 ) would become asunaprevir, named after its inventor Li-Qiang Sun.
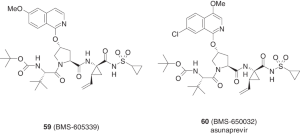
In our studies, the isoquinoline P 2 * moiety had offered improved PK properties, most effectively illustrated by the comparison between the data compiled in Table 2 for 61 and 62 [ 66 , 67 ] The plasma area under the curve (AUC) in rats for the isoquinoline 61 measured over 4 h post-dose was almost 700-fold higher than that for the matched quinoline 62 whilst the liver levels of 61 were 100-fold higher. This is a remarkable effect for a seemingly subtle structural change in a molecule with a molecular weight of ~750 Da and which is not reflected in the antiviral profiles. A second example in a matched molecular pair (MMP) analysis of P 1 -P 3 macrocycles amplified the difference to a 28,000-fold effect on the plasma area AUC and 1524 fold in liver exposure.
With the advent of the HCV replicon assay, which in its initial iteration was a GT-1b variant, the virology team engaged in a screening campaign that assessed the effects of ~200,000 compounds against HCV and a bovine viral diarrhea virus (BVDV) replicon as a phylogenetically closely-related Flavivirus that functioned as a stringent counterscreen with which to triage hits [ 68 ]. The thiazolidinone 63 was identified as the only lead inhibitor, with its uniqueness emphasized by the absence of additional effective replicon inhibitors following screening of another 800,000 compounds [ 69 ]. Thiazolidinone 63 was a member of a purely prospective library synthesized in-house that differed significantly from literature compounds which were typically benzylidene derivatives prepared by a Knoevenagel condensation reaction between an aldehyde and the core heterocycle. Resistance studies with 63 mapped to NS5A, a protein with no known enzymatic activity that, even after 20 years of study, remains enigmatic in nature [ 70 , 71 ]. While studies around the furan and fluorophenyl moieties gave a good dynamic range of potency, the SARs were less than precise and somewhat nebulous in nature in contrast to that associated with the alanine moiety, where there was high sensitivity to small molecular edits [ 68 , 72 ]. Thus, the D-Ala isomer 64 was 200-fold less potent than 63 while glycine ( 65 ) and proline ( 66 ) were the only other active representatives of naturally occurring amino acids, with D-proline also considerably less potent than the natural L-isomer 66 [ 72 ]. However, as our familiarity with this chemotype grew, we became aware that some of them were inherently unstable. Concern about precisely what we were working with was heightened when incubation of the 67 , a close but more considerably more potent analogue of 63 , in replicon media for several days, during which time the compound was completely degraded, remarkably did not diminish its antiviral potency. Larry Snyder’s determination to understand this phenomenon was well-placed and he engaged the help of John Leet, the last vestige of the BMS natural products isolation group. John conducted a biogram analysis of the media that had been incubated with 67 , which identified two very minor degradants that demonstrated antiviral activity in addition to the more prominent inactive compounds that we had already characterized. The experiment was repeated on a larger scale with the compound incubated at a concentration of 100 µM in 2 liters of media, which allowed isolation of 1.1 mg of each of the active degradants which were characterized as dimers 68 , with one converting to the more thermodynamically stable isomer when heated in DMSO at 50 °C in an NMR tube. We had determined that degradation of 67 in DMSO was the result of oxidation at C-5 of the thiazolidinone heterocycle to generate a radical intermediate, with products presumably formed by a reaction with molecular oxygen, a diradical in its ground state. The presence of small amounts of the dimers 68 suggested that the C-5 radical, which is stabilized in a captodative fashion by the C=O, sulfur and phenyl substituents, was sufficiently long-lived to be able to find another molecule in the cell culture media, no doubt facilitated by some aggregative association. Given the SAR observations, we postulated that the pharmacophore might be represented by the bibenzyl 69 , a compound readily accessible from the stilbene 70 which, in turn, was prepared from the commercially-available embedded stilbene diamine. This hypothesis turned out to be correct, with the bibenzyl derivative 69 a potent antiviral agent in the GT-1b replicon while the olefin 70 was even more active, with a remarkable 350-fold potency advantage over 69 [ 71 , 72 , 73 ]. As a consequence of this development, the crystal structure of the amino terminal domain of NS5A, which revealed a dimeric species, that was published almost exactly 3 years after our discovery held no surprises, although it did create many questions and ideas about the MOA of our compounds [ 74 ].
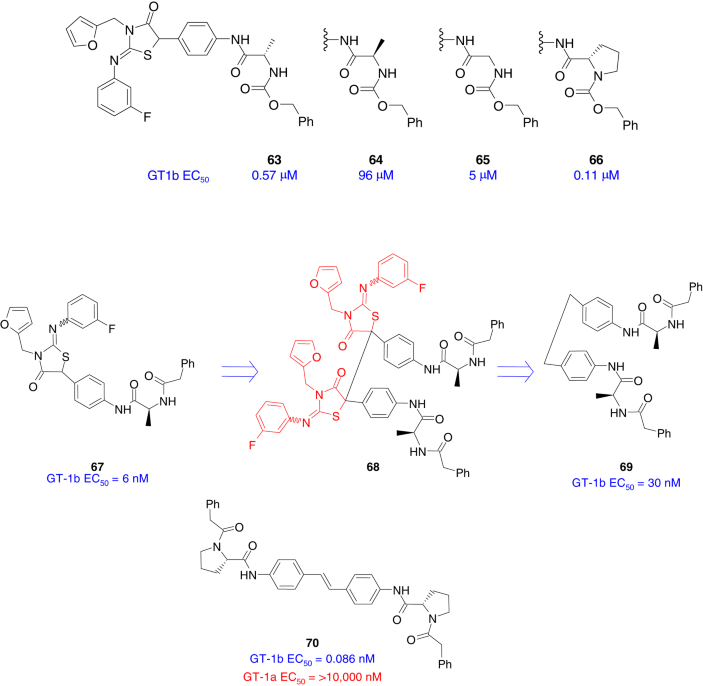
Whilst the discovery of 70 held promise, the olefin was subject to light-induced isomerization and concern was expressed about the two embedded aniline moieties that, given the peptide-like nature of the terminal elements, had the potential to be released in vivo; however, the more significant challenge came with the discovery that 70 was inactive toward a newly developed GT-1a replicon [ 71 ]. While this gave a clear focus to the subsequent optimization campaign, building in GT-1a inhibition proved to be an arduous enterprise, with management patience wearing thin at least twice over the almost 4-year period of study. However, each threat to terminate the program within a few weeks of giving notice coincided with developments in SARs that preserved the program. While management seemed to believe in a cause/effect relationship, the reality was that the chemists were toiling diligently, with the breakthroughs the result of the kind of commitment and attention to detail that we were accustomed to contributing. Daclatasvir ( 71 ) was conceived of and synthesized by Makonen Belema and Van Nguyen and ultimately became the first HCV NS5A replication complex inhibitor to enter clinical trials, which occurred in November of 2007 [ 69 , 71 ]. The Phase 1b clinical data with 71 were quite remarkable, with a dose of 1 mg associated with a 1.8 log 10 reduction viral load measured at 24 h post-dose while a single 100 mg dose exhibited higher efficacy, with a 3.6 log 10 reduction in plasma viremia measured at 48 h that persisted for 6 days [ 69 ]. There are some days that you never forget and seeing the initial efficacy associated with the 1 mg dose of 71 certainly qualifies, since it fully confirmed the translation of our preclinical discoveries to the clinical environment. That, and the subsequent development of the fuller graph presented in the article published in Nature in 2010 that describes the discovery of 71 , remain as indelible memories [ 69 ].
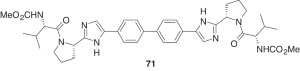
The NS3/4A inhibitor 60 and the NS5A inhibitor 71 were advanced into clinical trials within 2 months of each other which set the stage the for what would be described as a ground-breaking clinical trial in which a cohort of 11 non-responders to existing therapy were administered a combination of the two drugs for 24 weeks [ 75 ]. Two of the 11 patients were infected with GT-1b virus while the remaining nine were infected with the GT-1a subtype. A control group of 10 non-responders received the two small molecule drugs along with PEG-IFNα and ribavirin, the extant standard of care, for 24 weeks. In the dual combination cohort, four patients (the two GT-1b infected subjects and two who were infected with GT1a) remained virus free 12 weeks after the end of therapy, while all of the quadruple therapy group remained virus free at the same 12 week time point. This result demonstrated for the first time that an HCV infection could be cured by small molecule drugs in the absence of immune stimulation, an outcome described by Raymond T. Chung in the accompanying editorial as “A watershed moment in the treatment of hepatitis C” [ 76 ]. This result set the stage for the successful development of combinations of direct-acting antiviral agents (DAAs) that are curative after just 12 weeks of well tolerated therapy [ 77 ]. Indeed, just 2 years later the curing of chronic HCV infection was described as “the arc of a medical triumph” but since DAAs are all small molecule inhibitors, the accomplishment is perhaps better described as “the arc of a medicinal chemistry triumph” [ 78 ]. The impact of curative HCV therapeutics has manifested clinically as a significant reduction in the incidence of liver transplants attributable to the virus, with improvements in liver health and function and a reduction in mortality in those cured of the infection, a remarkably different circumstance to what had been predicted in the years before the advent of DAAs [ 79 , 80 , 81 , 82 , 83 ].
The success of the dual combination of 60 and 71 in curing the two GT-1b-infected patients in this clinical study led to the development of the two drugs being directed toward Japan where GT-1b was dominant, with marketing approval occurring on July 4th, 2014 [ 84 ].
An interesting aspect of the biochemical pharmacology of 71 is that of target vulnerability, defined as the fractional target occupancy required to produce a pharmacodynamic effect [ 85 ]. It was calculated that in GT-1b replicons, the ratio of HCV NS5A protein to 71 is of the order of 47,000:1 which equates to 23,500:1 based on the dimeric nature of the protein that is believed to be the drug target [ 86 ]. NS5A is and RNA binding protein that is believed to function in an oligomeric form in which a single molecule of 71 can disrupt the function of the higher order protein in the replication complex and during the packaging of RNA into the developing virion. The role of NS5A in RNA packaging was not captured in our replicon experiments but was shown in subsequent studies to contribute to the exceptionally rapid fall in plasma RNA observed in the clinical trials with 71 . In addition to highlighting the vulnerability of oligomeric viral proteins to therapeutic intervention, an examination of this mechanistic hypothesis led to the identification of molecules like 72 that were able to resensitize HCV that had developed resistance to 71 by binding to an allosteric site [ 86 ].
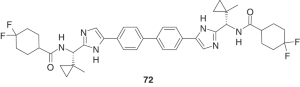
The final molecule in the HCV franchise was beclabuvir ( 73 ) which encountered some development challenges but was eventually approved in Japan on December 20th, 2016 as part of a fixed-dose triple combination with 60 and 70 referred to as DCV-TRIO and marketed under the tradename Xymency TM [ 87 , 88 ]. Interestingly, after presenting the discovery of 73 at the first-time disclosures session at the Spring meeting of the “American Chemical Society” held in San Diego in March 2012, where I stepped in for John Kadow who was not able to travel, the next day we found ourselves the subject of commentary by Derek Lowe in his blog [ 89 , 90 ]. The title of the blog entry was “What’s the ugliest drug? Or the ugliest drug candidate?” and 73 was specifically highlighted along with additional comments on 71 (see Box 1 ). The discovery and development of HCV inhibitors advanced acceptable drug properties beyond the rule of 5 guidelines whilst adding to an evolving complexity of drug candidates and the discovery of 73 had been particularly challenging. Nevertheless, the clinical success of HCV inhibitors has helped to set the stage for the contemporary era in which the pursuit of a wider range of modalities, including macrocyclic peptide drug candidates and targeted protein degraders, are further pushing the boundaries of drug design practices [ 91 , 92 , 93 , 94 ].
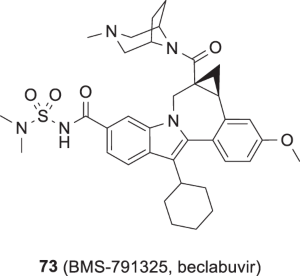
The final drug discovery vignette that I will share is focused on inhibitors of HIV-1 maturation. As we were winding down the HCV inhibitor programs, we reassessed the field of HIV-1 drug discovery and development where drugs had continued to be added to the pharmacopeia during the decade between 1998 and 2008 [ 95 ]. The landscape analysis concluded that there remained unmet medical need for new HIV-1 drugs acting by unique mechanisms that would reset the resistance clock. In addition, the field was beginning to consider approaches to enabling elite control over replication, where developments in immuno-oncology by our colleagues were germane, and cure of the disease, both of which remain significant challenges [ 96 , 97 ]. We had been monitoring the fate of the HIV-1 maturation inhibitor bevirimat ( 74 ), a mechanistically interesting acylated derivative of betulinic acid, for some time following an earlier interest in the molecule as a potential licensing candidate [ 98 , 99 ]. In Phase 2 clinical trials, 74 had demonstrated dichotomous efficacy, with a 50% response rate that was shown to be due to the presence of pre-existing mutations in the virus Gag protein, an analysis that was published coincident with our rekindled interest [ 100 ]. These observations allowed for the development of a relatively straightforward screening tier to assess virus susceptibility but, a priori, we had no understanding of whether or not this was a solvable problem and our experience with enhancing the spectrum of action of antiviral activity of small molecule drugs had been mixed [ 101 ]. Bevirimat ( 74 ) had been discovered using a phenotypic screen and although a potent antiviral agent in cell culture, suffered from a 100-fold serum effect and exhibited sub-optimal solubility problems that had plagued its development. The clinical challenges encountered by 74 presumably contributed to its attracting only limited attention by the pharmaceutical industry but we felt that there was ample opportunity for a more detailed SAR study beyond the existing work which, at that time, we considered to be rudimentary in nature. Our initial focus was on understanding the relationship between the carboxylate moiety appended to the C-3 OH of 74 and the triterpenoid core. In a remarkable turn of events, the very first compound that Alicia Regueiro-Ren and her team prepared was the C-3-benzoic acid derivative 75 which exhibited in vitro potency and in vivo PK properties in the rat that were comparable to 74 [ 101 ]. Adding to the luster of 75 , the effect of added human serum albumin on the antiviral potency was 10-fold lower than that experienced by 74 . With this motif in hand, modification at the C-20 acid moiety, the only other convenient handle in the molecule, was pursued. A broad SAR survey was conducted at this site which revealed that mildly basic amines in this region of the pharmacophore conferred the targeted antiviral profile, including encompassing clinically-relevant and challenging polymorphic viruses, whilst the presence of carboxylic acid moieties was associated with enhanced oral exposure. Guided by these principles, a thiomorpholine dioxide was identified as a mildly basic amine in which the sulfone element was interpreted as mimicking the oxygen atoms of a carboxylic acid moiety but without the burden of charge; thus, this ring system was viewed as a bioisostere of glycine or β-alanine [ 101 ]. The final molecular edit was to conduct a Curtius rearrangement on the C-20 acid which installed an amine attachment element attached directly to the core where the basicity was shielded, taking a cue from SAR observations where this concept led to an enhanced PK profile. This resulted in the identification of BMS-955176 ( 76 ), which was subsequently labeled as GSK3532795 following its acquisition by ViiV Healthcare, as a molecule meeting targeted antiviral and PK criteria that was advanced into clinical study in May, 2012 [ 101 ].

As monotherapy administered over 10 days to HIV-1 infected patients, 76 was effective at reducing viral load and fully suppressed the polymorphic viruses that had complicated the development of 74 . However, clinical development of 76 had to be abandoned following a Phase 2b study in which it was administered in combination with the nucleoside analogues tenofovir and emtricitabine, as a consequence of gut intolerability and the emergence of resistant virus that was often a complex complexion involving multiple mutations [ 102 ]. At the time that 76 was advanced, the back-up program had focused on further enhancement of the antiviral profile, which we considered a reasonable objective in the absence of a known liability. This proved to be prescient and by the time the clinical data with 76 had become clear, we were well advanced on the path to identifying fiprivirimat ( 77 ) [ 103 ]. The approach adopted was to screen molecules with C-3 variants against the key emerging clinical mutations which identified two series with promising activity, one that used the bicyclobutane found in 78 as an interesting phenyl bioisostere and a second that was based on a cyclohexene chemotype and exemplified by 79 [ 103 ] The latter offered the better antiviral profile toward a broader range of clinical mutants and was optimized to 77 which is currently in Phase 2 clinical trials where it has demonstrated dose-related reductions in viral load in HIV-1-infected subjects following oral administration on a once daily dosing regimen [ 103 , 104 , 105 ].
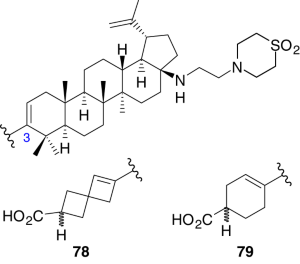
HIV-1 maturation inhibitors bind to the viral capsid-SP1 protein and interfere with protease-mediated cleavage to release the SP1 peptide, the final and rate-determining step in maturation [ 106 ]. The capsid protein comprises of 215–250 hexamers interspersed with precisely 12 pentamers that confer the unique cone shape to the assembled capsid. As an oligomeric structure, inhibition of capsid protein processing has been shown to exert a dominant negative effect on capsid uncoating, which is a carefully choreographed event, such that the incorporation of <5% of defective proteins is sufficient to exert an antiviral effect [ 107 , 108 , 109 ]. Thus, interfering with HIV-1 capsid maturation also represents a target of high vulnerability [ 85 ].
Box 1. Narrative on the ACS presentation on the invention of beclabuvir ( 76 ) by Derek Lowe in his blog
What’s the Ugliest Drug? Or The Ugliest Drug Candidate?
“What’s still making its way through the clinic can be even stranger-looking. Some of the odder candidates I’ve seen recently have been for the hepatitis C proteins NS5A and NS5B. Bristol Myers Squibb has disclosed some eye-openers, such as BMS-790052. (To be fair, that target seems to really like chemical matter like this, and the compound, last I heard, was moving along through the clinic.)
And yesterday, as Carmen Drahl reported from the ACS meeting in San Diego, the company disclosed the structure of BMS-791325, a compound targeting NS5B. That’s a pretty big one, too - the series it came from started out reasonably, then became not particularly small, and now seems to have really bulked up, and for the usual reasons - potency and selectivity. But overall, it’s a clear example of the sort of “compound bloat” that overtakes projects as they move on.”
Derek Lowe “ In the Pipeline ” blog, March 26th, 2012
In a 40-year career in drug discovery and development, I have witnessed tremendous change, the vast majority of which has been for the good and has significantly enhanced the process of candidate identification and in vitro and in vivo profiling. In the early days of my career, we did not enjoy the kind of preclinical PK support or liability profiling that is commonplace today and which is critical to the identification of quality clinical compounds. The changes implemented over the last four decades have subtended the discovery of many innovative transformational medicines and small molecule drugs continue to exert a significant impact on human health and longevity, although in a way that is not always fully appreciated by the public at large [ 81 , 110 , 111 , 112 , 113 ]. The effects of small molecule drugs on mortality associated with an HIV-1 infection has been substantial whilst the curing of HCV is a significant achievement, although the handful of successfully marketed HCV therapies reflect a rather dismal success rate of just 2% [ 81 , 112 , 114 ].
I joined the pharmaceutical industry during the ascent of CADD and experienced the advent of combinatorial chemistry in response to the emergence of high-throughput screening (HTS) both of which debuted with expectations that would prove to be somewhat unrealistic. However, both have found important roles in the drug discovery process and their historical evolution may provide guidance for the implementation and expectations of artificial intelligence. Nevertheless, synthetic organic chemistry remains a core competency in drug discovery and development, although in some respects this appears to have evolved into somewhat of a commodity item with the expansion of outsourcing and the commercial availability of a wide range of building blocks and capping agents that can facilitate rapid and convenient target compound assembly [ 115 , 116 , 117 ]. I believe that we fully embraced belief in the importance of synthetic acumen, viewing each synthetic challenge in the pursuit of answering biochemical pharmacological questions as a stimulus to invent new reagents and methodologies [ 6 , 22 , 23 , 55 ]. Physical organic chemistry is an inextricably critical component in understanding physicochemical properties and molecular recognition and, by extension, the principles of drug design [ 118 , 119 , 120 ]. This is an important principle which we pursued with vigor, with a number of the observations and discoveries forming the basis of Perspective and review articles that had the dual purpose of embellishing our knowledge whilst sharing our thoughts and insights with the broader medicinal chemistry community at large [ 121 , 122 , 123 , 124 , 125 , 126 , 127 , 128 , 129 , 130 , 131 , 132 , 133 ]. Indeed, although these endeavors were time-consuming, I encouraged my team to engage in communicating their scientific discoveries and we found this to be an immensely rewarding act of self-education and enlightenment that further enhanced our medicinal chemistry skills while garnering recognition from the broader medicinal chemistry community and burnishing the reputation of BMS. Notably, we were to learn that we were embracing words of wisdom from Bill Greenlee and other medicinal chemistry luminaries that we admire [ 134 , 135 , 136 ]. Several of these publications had a remarkable but unintended effect on my visibility within the medicinal chemistry community, most notably a synopsis on applications of bioisosteres in drug design [ 127 ]. I had a assembled a slide deck on this topic for an ACS Prospectives meeting held in Philadelphia in 2009 where Paul S. Anderson, the retired Senior Vice President of Chemical and Physical Sciences at DuPont Pharmaceuticals and the 2008 Priestley Medalist, was our plenary speaker. Paul had taken the book of slides home and e-mailed me a couple of weeks later to encourage me to synthesize the bioisostere material into a Perspective article for the Journal of Medicinal Chemistry . I had not contemplated that path but a query to Bill Greenlee was met with enthusiasm and the article was published in early 2011 [ 133 , 136 ]. The publication of this Perspective appeared to restore my standing with Derek Lowe, who commented on the article in his blog on March 22nd, 2011 (see Box 2 ), whilst also leading to invitations to present lectures on the topic at the annual “Drew Residential School on Medicinal Chemistry and Biology in Drug Discovery” and the biennial “Swiss Course on Medicinal Chemistry” [ 136 ]. These have been enjoyable experiences where I have been able to contribute to helping the next generation of medicinal chemists learn about what for me has been an enjoyable and rewarding career choice. These events have also created the opportunity to meet some wonderful scientists from all over the world who have become good friends and acquaintances.
I have had the good fortune to report to excellent supervisors, (in chronological order) Dr. John E. Lawson, J.J. Kim Wright, Graham Johnson, Carl P. Decicco, Joel C. Barrish, Percy H. Carter and Gregory D. Vite, who provided the kind of guidance and mentorship that allowed us ample opportunity to pursue our ideas although all were sources of good scientific suggestion that advanced our studies. They also acted as important sounding boards for discussion on a range of topics and I thank all of them for their support and collegiality. In prosecuting the individual programs, I was also fortunate to be able to collaborate with very many remarkable scientists, all of whom were creative and committed, with almost all of them co-authors on the publications that describe the many scientific discoveries that we made together and shared with the broader drug discovery community. We pursued many first-in-class targets with a strong communal belief that we would be successful and I particularly want to express my gratitude to them for their collaborative spirit, creativity and commitment which made the challenges associated with each discovery campaign more enjoyable and the achievements all the more rewarding. This was particularly the case in virology where we relied extensively on phenotypic screening which delivered antiviral leads with novel MOAs that we could not have anticipated and many of which would be very difficult to reduce to a biochemical screen. The HCV NS5A inhibitor campaign elegantly exemplifies that discovery principle, with this viral protein function still unapproachable using a biochemical assay. I believe that you discover what you screen for, so screen design is of paramount importance and the high content nature of a phenotypic screen maximizes the chance of success whilst also identifying novel MOAs that can be challenging to anticipate.
Every drug needs a champion and individual bias can influence a discovery and development portfolio. Fortunately, Carl Decicco, my supervisor through much of the successful HCV inhibitor campaigns, believed both in what we were pursuing and in our ability to deliver drug candidates. These programs were not always viewed favorably in the many corners of BMS although, not surprisingly, the Phase 2a clinical efficacy data changed views considerably. Carl and Rich Colonno were strong advocates for the preclinical HCV work, no doubt a reflection of their own direct experiences with the challenge of discovering effective and potent antiviral agents. This is the kind of intimate experience that can give a manager a stronger sense and understanding of the challenges faced by those designing and synthesizing molecules in the pursuit of a meaningful drug candidate. However, the effect extends beyond the manifestation of empathy and support and I’ve learned several times that selecting a target and pursuing it with a strong sense of conviction in its potential can be of paramount importance to success. However, decision making in drug discovery and development is a delicate balancing act, inherently flawed based on an absence of predictive accuracy, and knowing when to conclude a discovery program with grace is also an important trait. During my career, several substantial opportunities were missed that were subsequently exploited by others, notably PGI 2 partial agonists, CFTR modulators and RSV fusion inhibitors. However, the science that we conducted and the molecules and pharmacophores that we defined have been of benefit to mankind, which is ultimately a source of satisfaction.
The effects of small molecules can be remarkable, illustrated most effectively by 71 which addresses a target that would be deemed undruggable in a world where reductionist screening has become a key focus. I believe that the future of small molecule drugs has never been brighter, with proximity-induced protein and RNA modulation emerging technologies that have immense potential, and the rising appreciation of innovative screen design. While these technologies will have their successes, for some there will be setbacks along the way. However, that is the hallmark of drug discovery and development, an arduously challenging enterprise that does reward those with fortitude and which translates into the transformative medicines that society needs for many challenging diseases.
Box 2. Narrative by Derek Lowe in his blog on the Perspective article “Synopsis of some recent tactical application of bioisosteres in drug design.” Published in the Journal of Medicinal Chemistry in 2011 [ 133 , 136 ]
A 200-Proof Shot of Medicinal Chemistry
For the chemists out there in the crowd: have you been looking for a paper to read that’s filled, beginning to end, with good, solid, old-fashioned medicinal chemistry? Look no further than this one, on recent reports of isosteres. This sort of thing is still the heat of med-chem as it’s practiced in the real world—messing around with the structure of an active molecule to see what you can improve and what you can get away with.
If you’re not a medicinal chemist, the idea of a bioisostere is some chemical group that can substitute for another one. Classic examples are things like swapping in a tetrazole ring for a carboxylic acid or an oxadiazole for an ester. Here are some examples—even if your organic chemistry is shaky, you can see the similarities across these structures. If it works, you can change the other properties of your molecule (solubility, stability, selectivity) for the better while still keeping the key features that made the original group valuable for activity. It’s not something that just automatically comes through every time—sometimes there just is no substitute - but it works enough of the time to be one of the essential techniques.
Abbreviations
acyl-CoA:cholesterol acyltransferase
attachment inhibitor
area under the curve
bis-in-die (twice daily)
Bristol Myers Squibb
blood platelet aggregation
bovine viral diarrhea virus
computer-aided drug design
cyclic adenosine monophosphate
cystic fibrosis transmembrane conductance regulator
central nervous system
Cambridge Structural Database
cardiovascular
U.S. Food and Drug Administration
gastrointestinal
hemagglutinin
hepatitis B virus
hepatitis C virus
human immunodeficiency virus 1
high-throughput screening
investigational new drug
intravenous
matched molecular pair
mechanism of action
normal healthy volunteer
nuclear magnetic resonance
non-steroidal anti-inflammatory drug
phosphodiesterase
prostacyclin
proof-of-concept
pharmacokinetic
ribonucleic acid
respiratory syncytial virus
structure-activity relationship
Schlafen 12
sinoatrial node recovery time
United States
United States Adopted Names
Brown PJ, Jones DN, Khan MA, Meanwell NA, Richards PJ. Conjugate additions to α,β-unsaturated sulphoxides: syntheses of cyclopentenones and 9-deoxyprostanoids. J Chem Soc Perkin Trans I. 1984:2049–60. https://doi.org/10.1039/P19840002049 .
Johnson CR, Meanwell NA. Ketone methylenation with optical resolution. Total synthesis of the ginseng sesquiterpene (-)-β-panasinsene and its enantiomer. J Am Chem Soc. 1981;103:7667–9. https://doi.org/10.1021/ja00415a052 .
Article CAS Google Scholar
Johnson CR, Bis KG, Cantillo JH, Meanwell NA, Reinhard MFD, Zeller JR, et al. Preparation and reactions of sulfonimidoyl fluorides. J Org Chem. 1983;48:1–3. https://doi.org/10.1021/jo00149a001 .
Meanwell NA. Anagrelide—a clinically effective cAMP phosphodiesterase 3A inhibitor with molecular glue properties. ACS Med Chem Lett. 2023;14:350–61. https://doi.org/10.1021/acsmedchemlett.3c00092 .
Article CAS PubMed Google Scholar
Meanwell NA, Roth HR, Smith Edward CR, Wedding DL, Wright JJK, Fleming JS, et al. 1,3-Dihydro-2 H -imidazo[4,5- b ]quinolin-2-ones—inhibitors of blood platelet cAMP phosphodiesterase and induced aggregation. J Med Chem. 1991;34:2906–16. https://doi.org/10.1021/jm00113a034 .
Meanwell NA, Roth HR, Smith ECR, Wedding DL, Wright JJK. Diethyl 2,4-dioxoimidazolidine-5-phosphonates: Horner-Wadsworth-Emmons reagents for the mild and efficient preparation of C-5 unsaturated hydantoin derivatives. J Org Chem. 1991;56:6897–904. https://doi.org/10.1021/jo00024a036 .
Meanwell NA, Pearce BC, Roth HR, Smith ECR, Wedding DL, Wright JJK, et al. Inhibitors of blood platelet cAMP phosphodiesterase. 2. Structure-activity relationships associated with 1,3-dihydro-2 H -imidazo[4,5- b ]quinolin-2-ones substituted with functionalized side chains. J Med Chem. 1992;35:2672–87.
Meanwell NA, Dennis RD, Roth HR, Rosenfeld MJ, Smith ECR, Wright JJK, et al. Inhibitors of blood platelet cAMP phosphodiesterase. 3. 1,3-Dihydro-2 H -imidazo[4,5- b ]quinolin-2-one derivatives with enhanced aqueous solubility. J Med Chem. 1992;35:2688–96.
Lautenschlager HH, Prop G, Niemann R. Octimibate sodium. Drugs Future. 1986;11:26–7.
Article Google Scholar
Seiler S, Brassard CL, Arnold AJ, Meanwell NA, Fleming JS, Keely SL Jr. Octimibate inhibition of platelet aggregation: stimulation of adenylate cyclase through prostacyclin receptor activation. J Pharm Exp Ther. 1990;255:1021–6. http://jpet.aspetjournals.org/content/255/3/1021 .
CAS Google Scholar
Raju TN. The Nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918-99), and George H Hitchings (1905-98). Lancet. 2000;355:1022. https://doi.org/10.1016/S0140-6736(05)74775-9 .
Meanwell NA, Rosenfeld MJ, Trehan AK, Wright JJK, Brassard CL, Buchanan JO, et al. Nonprostanoid prostacyclin mimetics. 2. 4,5-diphenyloxazole derivatives. J Med Chem. 1992;35:3483–97. https://doi.org/10.1021/jm00097a006 .
Meanwell NA, Rosenfeld MJ, Wright JJK, Brassard CL, Buchanan JO, Federici ME, et al. Structure-activity relationships associated with 3,4,5-triphenyl-1 H -pyrazole-1-nonanoic acid, a nonprostanoid prostacyclin mimetic. J Med Chem. 1992;35:389–97. https://doi.org/10.1021/jm00080a028 .
Meanwell NA, Romine JL, Rosenfeld MJ, Martin SW, Trehan AK, Wright JJK, et al. Nonprostanoid prostacyclin mimetics. 5. Structure-activity relationships associated with [3-[4-(4,5-diphenyl-2-oxazolyl)-5-oxazolyl]phenoxy]acetic acid. J Med Chem. 1993;36:3884–903. https://doi.org/10.1021/jm00076a018 .
Meanwell NA, Romine JL, Seiler SM. Non-prostanoid prostacyclin mimetics. Drugs Future. 1994;19:361–85.
Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6. https://doi.org/10.1021/jm901241e .
Nakamura A, Yamada T, Asaki T. Synthesis and evaluation of N-acylsulfonamide and N-acylsulfonylurea prodrugs of a prostacyclin receptor agonist. Bioorg Med Chem. 2007;15:7720–5. https://doi.org/10.1016/j.bmc.2007.08.052 .
Tran T-A, Kramer B, Shin Y-J, Vallar P, Boatman PD, Zou N, et al. Discovery of 2-(((1 R ,4 R )-4-(((4-chlorophenyl)(phenyl)carbamoyl)oxy)methyl)cyclohexyl)methoxy)acetate (ralinepag): an orally active prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. J Med Chem. 2017;60:913–27. https://doi.org/10.1021/acs.jmedchem.6b00871 .
Gribkoff VK, Starrett JE Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, et al. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med. 2001;7:471–7. https://doi.org/10.1038/86546 .
Macdonald JE, Poindexter GS. An anomalous metalation of a trimethylsilyl group. Tetrahedron Lett. 1987;28:1851–2. https://doi.org/10.1016/S0040-4039(00)95991-4 .
Creary X. Reaction of organometallic reagents with ethyl trifluoroacetate and diethyl oxalate. Formation of trifluoromethyl ketones and α-keto esters via stable tetrahedral adducts. J Org Chem. 1987;52:5026–30. https://doi.org/10.1021/jo00231a036 .
Hewawasam P, Meanwell N. A general method for the synthesis of isatins: preparation of regiospecifically functionalized isatins from anilines. Tetrahedron Lett. 1994;35:7303–6. https://doi.org/10.1016/0040-4039(94)85299-5 .
Meanwell NA, Sit S-Y, Gao J, Wong HS, Gao Q, St. Laurent DR, et al. Regiospecific functionalization of 1,3-dihydro-2 H -benzimidazol-2-one and structurally related cyclic urea derivatives. J Org Chem. 1995;60:1565–82. https://doi.org/10.1021/jo00111a014 .
Meanwell NA, Sit S-Y, Gao J, Boissard CG, Lum-Ragan J, Dworetzky SI, et al. N-Benzylated benzimidazol-2-one derivatives: activators of large-conductance Ca2 +- dependent K + channels. Bioorg Med Chem Lett. 1996;6:1641–6. https://doi.org/10.1016/0960-894X(96)00296-X .
Hewawasam P, Meanwell NA, Gribkoff VK, Dworetzky SI, Boissard CG. Discovery of a novel class of BK channel openers: enantiospecific synthesis and BK channel opening activity of 3-(5-chloro-2-hydroxyphenyl)-1,3-dihydro-3-hydroxy-6-(trifluoromethyl)-2 H -indol-2-ones. Bioorg Med Chem Lett. 1997;7:1255–60. https://doi.org/10.1016/S0960-894X(97)00202-3 .
Hewawasam P, Erway M, Moon SL, Knipe J, Weiner H, Boissard CG, et al. Synthesis and structure-activity relationships of 3-aryloxindoles: a new class of calcium-dependent, large conductance potassium (maxi-K) channel openers with neuroprotective properties. J Med Chem. 2002;45:1487–99. https://doi.org/10.1021/jm0101850 .
Hewawasam P, Gribkoff VK, Pendri Y, Dworetzky SI, Meanwell NA, Martinez E, et al. The synthesis and characterization of BMS-204352 (MaxiPost) and related 3-fluorooxindoles as openers of maxi-K potassium channels. Bioorg Med Chem Lett. 2002;12:1023–26. https://doi.org/10.1016/S0960-894X(02)00101-4 .
Gillman KW, Hewawasam P, Schmitz WD, Lopez OD, Starrett JE, Provencal DP. Preparation of phosphate prodrugs of fluorooxindoles. World Patent Appl. WO2003/080047 A1. 2003.
Romine JL, Martin SW, Meanwell NA, Gribkoff VK, Boissard CG, Dworetzky SI, et al. 3-[(5-Chloro-2-hydroxyphenyl)methyl]-5-[4-(trifluoromethyl)phenyl]-1,3,4-oxadiazol-2(3 H )-one, BMS-191011: opener of large-conductance Ca 2+ -activated potassium (maxi-K) channels, identification, solubility, and SAR. J Med Chem. 2007;50:528–42.
Hewawasam P, Ding M, Chen N, King D, Knipe J, Pajor L, et al. Synthesis of water-soluble prodrugs of BMS-191011: a maxi-K channel opener targeted for post-stroke neuroprotection. Bioorg Med Chem Lett. 2003;13:1695–8.
Hewawasam P, Erway M, Thalody G, Weiner H, Boissard CG, Gribkoff VK, et al. The synthesis and structure-activity relationships of 1,3-diaryl 1,2,4-(4 H )-triazol-5-ones: a new class of calcium-dependent, large conductance, potassium (maxi-K) channel opener targeted for urge urinary incontinence. Bioorg Med Chem Lett. 2002;12:1117–20.
Vrudhula VM, Dasgupta B, Qian-Cutrone J, Kozlowski ES, Boissard CG, Dworetzky SI, et al. Atropisomeric 3-(β-hydroxyethyl)-4-arylquinolin-2-ones as maxi-K potassium channel openers. J Med Chem. 2007;50:1050–7.
Chen X, Wang K. The fate of medications evaluated for ischemic stroke pharmacotherapy over the period 1995-2015. Acta Pharm Sin B. 2016;6:522–30. https://doi.org/10.1016/j.apsb.2016.06.013 .
Article PubMed PubMed Central Google Scholar
Gribkoff VK, Champigny G, Barbry P, Dworetzky SI, Meanwell NA, Lazdunski M. The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J Biol Chem. 1994;269:10983–6.
Bardin E, Pastor A, Semeraro M, Golec A, Hayes K, Chevalier B, et al. Modulators of CFTR. Updates on clinical development and future directions. Eur J Med Chem. 2021;213:113195.
Luo G, Torri A, Harte WE, Danetz S, Cianci C, Tiley L, et al. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J Virol. 1997;71:4062–70.
Article CAS PubMed PubMed Central Google Scholar
Yu K-L, Ruediger E, Luo G, Cianci C, Danetz S, Tiley L, et al. Novel quinolizidine salicylamide influenza fusion inhibitors. Bioorg Med Chem Lett. 1999;9:2177–80.
Deshpande MS, Wei J, Luo G, Cianci C, Danetz S, Torri A, et al. An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg Med Chem Lett. 2001;11:2393–6.
Yu K-L, Torri AF, Luo G, Cianci C, Grant-Young K, Danetz S, et al. Structure-activity relationships for a series of thiobenzamide influenza fusion inhibitors derived from 1,3,3-trimethyl-5-hydroxy-cyclohexylmethylamine. Bioorg Med Chem Lett. 2002;12:3379–82.
Wilber R, Kreter B, Bifano M, Danetz S, Lehman-McKeeman L, Tenney DJ, et al. Discovery and development of entecavir. In: Kazmierski WM, editor. Antiviral drugs. Hoboken, New Jersey: John Wiley & Sons; 2011. p. 401–16.
Bold G, Fässler A, Capraro H-G, Cozens R, Klimkait T, Lazdins J, et al. New aza-dipeptide analogs as potent and orally absorbed HIV-1 protease inhibitors: candidates for clinical development. J Med Chem. 1998;41:3387–401. https://doi.org/10.1021/jm970873c .
Yu K-L, Zhang Y, Civiello RL, Kadow KF, Cianci C, Krystal M, et al. Fundamental structure-activity relationships associated with a new structural class of respiratory syncytial virus inhibitor. Bioorg Med Chem Lett. 2003;13:2141–4.
Cianci C, Langley DR, Dischino DD, Sun Y, Yu K-L, Stanley A, et al. Targeting a binding pocket within the trimer-of-hairpins: small-molecule inhibition of viral fusion. Proc Natl Acad Sci USA. 2004;101:15046–51.
Wang T, Zhang Z, Wallace OB, Deshpande M, Fang H, Yang Z, et al. Discovery of 4-benzoyl-1-[(4-methoxy-1 H -pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2-( R )-methylpiperazine (BMS-378806): a novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J Med Chem. 2003;46:4236–9.
Lin P-F, Blair W, Wang T, Spicer T, Guo Q, Zhou N, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci USA. 2003;100:11013–8.
Cianci C, Genovesi EV, Lamb L, Medina I, Yang Z, Zadjura L, et al. Oral efficacy of a respiratory syncytial virus inhibitor in rodent models of infection. Antimicrob Agents Chemother. 2004;48:2448–54.
Vendeville S, Tahri A, Hu L, Demin S, Cooymans L, Vos A, et al. Discovery of 3-({5-chloro-1-[3-(methylsulfonyl)propyl]-1 H -indol-2-yl}methyl)-1-(2,2,2-trifluoroethyl)-1,3-dihydro-2 H -imidazo[4,5- c ]pyridin-2-one (JNJ-53718678), a potent and orally bioavailable fusion inhibitor of respiratory syncytial virus. J Med Chem. 2020;63:8046–58.
Stevens M, Rusch S, DeVincenzo J, Kim Y-I, Harrison L, Meals EA, et al. Antiviral activity of oral JNJ-53718678 in healthy adult volunteers challenged with respiratory syncytial virus: a placebo-controlled study. J Infect Dis. 2018;218:748–56.
Martinon-Torres F, Rusch S, Huntjens D, Remmerie B, Vingerhoets J, McFadyen K, et al. Pharmacokinetics, safety, and antiviral effects of multiple doses of the respiratory syncytial virus (RSV) fusion protein inhibitor, JNJ-53718678, in infants hospitalized with RSV infection: a randomized phase 1b study. Clin Infect Dis. 2020;71:e594–603.
Cockerill GS, Angell RM, Bedernjak A, Chuckowree I, Fraser I, Gascon-Simorte J, et al. Discovery of sisunatovir (RV521), an inhibitor of respiratory syncytial virus fusion. J Med Chem. 2021;64:3658–76.
DeVincenzo J, Tait D, Efthimiou J, Mori J, Kim Y-I, Thomas E, et al. A randomized, placebo-controlled, respiratory syncytial virus human challenge study of the antiviral efficacy, safety, and pharmacokinetics of RV521, an inhibitor of the RSV-F protein. Antimicrob Agents Chemother. 2020;64:e01884.
Pfizer Press Release. Pfizer completes acquisition of ReViral. 2022. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-completes-acquisition-reviral .
Meanwell NA, Krystal MR, Nowicka-Sans B, Langley DR, Conlon DA, Eastgate MD, et al. Inhibitors of HIV-1 attachment: the discovery and development of temsavir and its prodrug fostemsavir. J Med Chem. 2018;61:62–80.
Wang T, Kadow JF, Meanwell NA. Innovation in the discovery of the HIV-1 attachment inhibitor temsavir and its phosphonooxymethyl prodrug fostemsavir. Med Chem Res. 2021;30:1955–80.
CAS PubMed PubMed Central Google Scholar
Timmins P, Brown J, Meanwell NA, Hanna GJ, Zhu L, Kadow JF. Enabled clinical use of an HIV-1 attachment inhibitor through drug delivery. Drug Discov Today. 2014;19:1288–93.
Kozal M, Aberg J, Pialoux G, Cahn P, Thompson M, Molina J-M,and The BRIGHTE Trial Team, et al. Fostemsavir in adults with multidrug-resistant HIV-1 infection. N Engl J Med. 2020;382:1232–43.
Gartland M, Cahn P, DeJesus E, Diaz RS, Grossberg R, Kozal M, et al. Week 96 genotypic and phenotypic results of the fostemsavir phase 3 BRIGHTE study in heavily treatment-experienced adults living with multidrug-resistant HIV-1. Antimicrob Agents Chemother. 2022;66. https://doi.org/10.1128/aac.01751-21 .
Markham A. Fostemsavir: first approval. Drugs. 2020;80:1485–90.
Herrera C, Harman S, Aldon Y, Rogers P, Armanasco N, Ziprin P, et al. The entry inhibitor DS003 (BMS-599793): a BMS-806 analogue, provides superior activity as a pre-exposure prophylaxis candidate. AIDS. 2021;35:1907–17.
Combrink KD, Gülgeze HB, Meanwell NA, Pearce BC, Zulan P, Bisacchi GS, et al. 1,2-Benzisothiazol-3-one 1,1-dioxide inhibitors of human mast cell tryptase. J Med Chem. 1998;41:4854–60.
Yeung K-S, Meanwell NA, Qiu Z, Hernandez D, Zhang S, McPhee F, et al. Structure-activity relationship studies of a bisbenzimidazole-based, Zn 2+ -dependent inhibitor of HCV NS3 serine protease. Bioorg Med Chem Lett. 2001;11:2355–9.
Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–90.
Scola PM, Wang A, Good AC, Sun L-Q, Combrink KD, Campbell JA, et al. Discovery and early clinical evaluation of BMS-605339, a potent and orally efficacious tripeptidic acylsulfonamide NS3 protease inhibitor for the treatment of hepatitis C virus infection. J Med Chem. 2014;57:1730–55.
Mecozzi S, Rebek J Jr. The 55% solution: a formula for molecular recognition in the liquid state. Chem Eur J. 1998;4:1016–22.
Scola PM, Sun L-Q, Wang AX, Chen J, Sin N, Venables BL, et al. The discovery of asunaprevir (BMS-650032), an orally efficacious NS3 protease inhibitor for the treatment of hepatitis C virus infection. J Med Chem. 2014;57:1730–52.
McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, et al. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob Agents Chemother. 2012;56:5387–96.
Lemm JA, O’Boyle DR II, Liu M, Nower PT, Colonno R, Deshpande MS, et al. Identification of hepatitis C virus NS5A inhibitors. J Virol. 2010;84:482–91.
Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100.
Ross-Thriepland D, Harris M. Hepatitis C virus 1 NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol. 2015;96:727–38.
Meanwell NA, Belema M. The discovery and development of daclatasvir: an inhibitor of the hepatitis C virus NS5A replication complex. Top Med Chem. 2019;32:27–55.
Romine JL, St. Laurent DR, Leet JE, Martin SW, Serrano-Wu MH, Yang F, et al. Inhibitors of HCV NS5A: from iminothiazolidinones to symmetrical stilbenes. ACS Med Chem Lett. 2011;2:224–9.
Lemm JA, Leet JE, O’Boyle DR II, Romine JL, Huang XS, Schroeder DR, et al. Discovery of potent hepatitis C virus NS5A inhibitors with dimeric structures. Antimicrob Agents Chemother. 2011;55:3795–802.
Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435:374–9.
Lok AS, Gardiner DF, Lawitz E, Martorell C, Everson GT, Ghalib R, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med. 2012;366:216–24.
Chung RT. A watershed moment in the treatment of hepatitis C. N Engl J Med. 2012;366:273–5.
Chung RT, Baumert TF. Curing chronic hepatitis C—the arc of a medical triumph. N Engl J Med. 2014;370:1576–8.
Meanwell NA. 2015 Philip S. Portoghese medicinal chemistry lectureship. Curing hepatitis c virus infection with direct-acting antiviral agents: the arc of a medicinal chemistry triumph. J Med Chem. 2016;59:7311–51.
Manns MP, Maasoumy B. Breakthroughs in hepatitis C research: from discovery to cure. Nat Rev Gastroenterol Hepatol. 2022;19:533–50. https://doi.org/10.1038/s41575-022-00608-8
Flemming JA, Kim WR, Brosgart CL, Terrault NA. Reduction in liver transplant wait-listing in the era of direct-acting antiviral therapy. Hepatology. 2017;65:804–12.
Meanwell NA, Ewing WR. In praise of remarkably powerful centamolecular therapeutic agents. ACS Med Chem Lett. 2019;10:1094–7.
Janjua NZ, Wong S, Abdia Y, Jeong D, Buller-Taylor T, Adu PA, et al. Impact of direct-acting antivirals for HCV on mortality in a large population-based cohort study. J Hepatol. 2021;75:1049–57. https://doi.org/10.1016/j.jhep.2021.05.028 .
Rein DB, Wittenborn JS, Weinbaum CM, Sabin M, Smith BD, Lesesne SB. Forecasting the morbidity and mortality associated with prevalent cases of pre-cirrhotic chronic hepatitis C in the United States. Dig Liver Dis. 2011;43:66–72.
Article PubMed Google Scholar
Poole RM. Daclatasvir + asunaprevir: first global approval. Drugs. 2014;74:1559–71.
Tonge PJ. Drug-target kinetics in drug discovery. ACS Chem Neurosci. 2018;9:29–39.
Sun J-H, O’Boyle DR II, Fridell RA, Langley DR, Wang C, Roberts SB, et al. Resensitizing daclatasvir-resistant hepatitis C variants by allosteric modulation of NS5A. Nature. 2015;527:245–8.
Gentles RG, Ding M, Bender JA, Bergstrom CP, Grant-Young K, Hewawasam P, et al. Discovery and preclinical characterization of the cyclopropylindolobenzazepine BMS-791325, A potent allosteric inhibitor of the hepatitis C virus NS5B polymerase. J Med Chem. 2014;57:1855–79. https://doi.org/10.1021/jm4016894 .
Zappulo E, Scotto R, Buonomo AR, Maraolo AE, Pinchera B, Gentile I. Efficacy and safety of a fixed dose combination tablet of asunaprevir + beclabuvir + daclatasvir for the treatment of Hepatitis C. Expert Opin Pharmacother. 2020;21:261–73. https://doi.org/10.1080/14656566.2019.1697674 .
Kadow JF, Gentles R, Ding M, Bender J, Bergstrom C, Grant-Young K, et al. Discovery of BMS-791325, an Allosteric NS5B Replicase Inhibitor for the Treatment of Hepatitis C. First time Disclosures Session at the 243rd National ACS Meeting and Exposition, San Diego CA, March 25–29th, MEDI 23, 2012.
Lowe D. What’s the ugliest drug? Or the ugliest drug candidate? 2012. https://www.science.org/content/blog-post/what-s-ugliest-drug-or-ugliest-drug-candidate .
Meanwell NA. Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem Res Toxicol. 2016;29:564–616. https://doi.org/10.1021/acs.chemrestox.6b00043 .
Li J, Eastgate MD. Current complexity: a tool for assessing the complexity of organic molecules. Org Biomol Chem. 2015;13:7164–76. https://doi.org/10.1039/C5OB00709G .
Eastgate M, Schmidt M, Fandrick K. On the design of complex drug candidate syntheses in the pharmaceutical industry. Nat Rev Chem. 2017;1:0016. https://doi.org/10.1038/s41570-017-0016 .
Ermondi G, Jimenez DG, Sebastiano MR, Caron G. Rational control of molecular properties is mandatory to exploit the potential of PROTACs as oral drugs. ACS Med Chem Lett. 2021;12:1056–60. https://doi.org/10.1021/acsmedchemlett.1c00298 .
Tseng A, Seet J, Phillips EJ. The evolution of three decades of antiretroviral therapy: challenges, triumphs and the promise of the future. Br J Clin Pharm. 2015;79:182–94. https://doi.org/10.1111/bcp.12403 .
Li JZ, Blankson JN. How elite controllers and posttreatment controllers inform our search for an HIV-1 cure. J Clin Invest. 2021;131:e149414. https://doi.org/10.1172/JCI149414 .
Deeks SG, Archin N, Cannon P, Collins S, Jones RB, de Jong MAWP, et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat Med. 2021;27:2085–98. https://doi.org/10.1038/s41591-021-01590-5 .
Kashiwada Y, Hashimoto F, Cosentino LM, Chen C, Garrett PE, Lee K-H. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J Med Chem. 1996;39:1016–7. https://doi.org/10.1021/jm950922q .
Martin DE, Salzwedel K, Allaway GP. Bevirimat: a novel maturation inhibitor for the treatment of HIV-1 infection. Antivir Chem Chemother. 2008;19:107–13.
Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, et al. Susceptibility of human immunodeficiency virus type 1 to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag spacer peptide 1. Antimicrob Agents Chemother. 2009;53:2185–8.
Regueiro-Ren A, Swidorski JJ, Liu Z, Chen Y, Sin N, Sit S-Y, et al. Design, synthesis, and SAR of C-3 benzoic acid, C-17 triterpenoid derivatives. Identification of the HIV-1 maturation inhibitor 4-((1 R ,3a S ,5a R ,5b R ,7a R ,11a S ,11b R ,13a R ,13b R )-3a-((2-(1,1-dioxidothiomorpholino)ethyl)amino)-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl)-2,3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a,13b-octadecahydro-1 H -cyclopenta[ a ]chrysen-9-yl)benzoic acid (GSK3532795, BMS-955176). J Med Chem. 2018;61:7289–313.
Morales-Ramirez J, Bogner JR, Molina J-M, Lombaard J, Dicker IB, Stock DA, et al. Safety, efficacy, and dose response of the maturation inhibitor GSK3532795 (formerly known as BMS-955176) plus tenofovir/emtricitabine once daily in treatment-naïve HIV-1-infected adults: week 24 primary analysis from a randomized Phase IIb trial. PLoS ONE. 2018;13:e0205368.
Regueiro-Ren A, Sit S-Y, Chen Y, Chen J, Swidorski JJ, Liu Z, et al. The discovery of GSK3640254, a next generation inhibitor of HIV-1 maturation. J Med Chem. 2022;65:11927–48.
JoshI SR, Fernando D, Igwe S, McKenzie L, Krishnatry AS, Halliday F, et al. Phase I evaluation of the safety, tolerability, and pharmacokinetics of GSK3640254, a next-generation HIV-1 maturation inhibitor. Pharm Res Perspect. 2020;8:e00671. https://doi.org/10.1002/prp2.671 .
Spinner CD, Felizarta F, Rizzardini G, Philibert P, Mitha E, Domingo P, et al. Phase IIa proof-of-concept evaluation of the antiviral efficacy, safety, tolerability, and pharmacokinetics of the next-generation maturation inhibitor GSK3640254. Clin Infect Dis. 2022;75:786–94. https://doi.org/10.1093/cid/ciab1065 .
Regueiro-Ren A, Dicker IB, Hanumegowda U, Meanwell NA. Second generation inhibitors of HIV-1 maturation. ACS Med Chem Lett. 2019;10:287–94.
Müller B, Anders M, Akiyama H, Welsch S, Glass B, Nikovics K, et al. HIV-1 gag processing intermediates transdominantly interfere with HIV-1 infectivity. J Biol Chem. 2009;284:29692–703.
Lee S-K, Harris J, Swanstrom R. A strongly transdominant mutation in the human immunodeficiency virus type 1 gag gene defines an Achilles heel in the virus life cycle. J Virol. 2009;83:8536–43.
Checkley MA, Luttge BG, Soheilian F, Nagashima K, Freed EO. The capsid-spacer peptide 1 Gag processing intermediate is a dominant-negative inhibitor of HIV-1 maturation. Virology. 2010;400:137–44.
Lichtenberg FR. Sources of U.S. longevity increase, 1960-2001. Quart Rev Econ Financ. 2004;44:369–89. https://doi.org/10.1016/j.qref.2004.05.005 .
Lichtenberg FR. The impact of new drug launches on longevity: evidence from longitudinal, disease-level data from 52 countries, 1982-2001. Int J Health Care Financ Econ. 2005;5:47–73. https://doi.org/10.1007/s10754-005-6601-7 .
Lichtenberg FR. The effect of new drug approvals on HIV mortality in the US, 1987-1998. Econ Hum Biol. 2003;1:259–66. https://doi.org/10.1016/S1570-677X(02)00031-X .
Hostenkamp G, Lichtenberg FR. The impact of recent chemotherapy innovation on the longevity of myeloma patients: US and international evidence. Soc Sci Med. 2015;130:162–171. https://doi.org/10.1016/j.socscimed.2015.02.003 .
Melnikova I. Hepatitis C therapies. Nat Rev Drug Discov. 2008;7:799–800. https://doi.org/10.1038/nrd2661 .
Campos KR, Coleman PJ, Alvarez JC, Dreher SD, Garbaccio RM, Terrett NK, et al. The importance of synthetic chemistry in the pharmaceutical industry. Science. 2019:eaat0805. https://doi.org/10.1126/science.aat0805 .
Blakemore DC, Castro L, Churcher I, Rees DC, Thomas AW, Wilson DM, et al. Organic synthesis provides opportunities to transform drug discovery. Nat Chem. 2018;10:383–94. https://doi.org/10.1038/s41557-018-0021-z .
Grygorenko OO, Volochnyuk DM, Ryabukhin SV, Judd DB. The symbiotic relationship between drug discovery and organic chemistry. Chem Eur J. 2020;26:1196–237. https://doi.org/10.1002/chem.201903232 .
Persch E, Dumele O, Diederich F. Molecular recognition in chemical and biological systems. Angew Chem Int Ed. 2015;54:3290–327. https://doi.org/10.1002/anie.201408487 .
Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–84. https://doi.org/10.1021/jm100112j .
Kuhn B, Gilberg E, Taylor R, Cole J, Korb O. How significant are unusual protein−ligand interactions? Insights from database mining. J Med Chem. 2019;62:10441–55. https://doi.org/10.1021/acs.jmedchem.9b01545 .
Bender JA, Meanwell NA, Wang T. The mono-functionalization of symmetrical polyamines. Tetrahedron. 2002;58:3111–3128.
Meanwell NA. Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol. 2011;24:1420–56.
Yeung K-S, Meanwell NA. Inhibitors of hERG channel trafficking—a cryptic mechanism for QT prolongation. Annu Rep. Med Chem. 2013;48:335–52.
Google Scholar
Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. Applications of fluorine in medicinal chemistry. J Med Chem. 2015;58:8315–59.
Meanwell NA. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J Med Chem. 2018;61:5822–80.
Johnson BM, Shu Y-Z, Zhuo X, Meanwell NA. Metabolic and pharmaceutical aspects of fluorinated compounds. J Med Chem. 2020;63:6315–86.
Subbaiah MAM, Meanwell NA. Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J Med Chem. 2021;64:14046–128.
Meanwell NA, Loiseleur O. Applications of isosteres of piperazine in the design of biologically active compounds. Part 1. J Agric Food Chem. 2022;70:10942–71.
Meanwell NA, Loiseleur O. Applications of isosteres of piperazine in the design of biologically active compounds. Part 2. J Agric Food Chem. 2022;70:10972–1004.
Beno BR, Yeung K-S, Bartberger MD, Pennington LD, Meanwell NA. A survey of the role of non-covalent sulfur interactions in drug design. J Med Chem. 2015;58:4383–438.
Wu Y-J, Meanwell NA. Geminal diheteroatomic motifs: some applications of acetals, ketals and their sulfur and nitrogen homologues in medicinal chemistry and drug design. J Med Chem. 2021;64:9786–874.
Greenlee WJ. Adventures in medicinal chemistry: a career in drug discovery. Annu Rep Med Chem. 2014;19:11–23.
Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–91.
Hatley RJD, Procopiou PA, McLachlan SP, Westendorf LE, Meanwell NA, Ewing WR, et al. Writing your next medicinal chemistry article: journal bibliometrics and guiding principles for industrial authors. J Med Chem. 2020;63:14336–56. https://doi.org/10.1021/acs.jmedchem.0c01159 .
Murcko MA. What makes a great medicinal chemist? A personal perspective. J Med Chem. 2018;61:7419–24. https://doi.org/10.1021/acs.jmedchem.7b01445 .
Lowe D. A 200-proof shot of medicinal chemistry. In the Pipeline. 2011. https://www.science.org/content/blog-post/200-proof-shot-medicinal-chemistry .
Download references
Acknowledgements
I wish to thank my late parents Jack and Marjorie for instilling in me their values and work ethics, my wife Patricia and my children, Emily and Stephen, for their enduring support without which a career would not have been possible. I would also like to express my gratitude to Professor Longqin Hu, Editor in Chief of Medicinal Chemistry Research , for his suggestion of developing a special issue of the Journal that this article appears in and to my colleagues from BMS, Drs. John Kadow, Kap-Sun Yeung and Murali Dhar, for assuming the role of editors for the issue.
Author information
Authors and affiliations.
The Baruch S. Blumberg Institute, 3805 Old Easton Road, Doylestown, PA, 18902, USA
Nicholas A. Meanwell
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Nicholas A. Meanwell .
Ethics declarations
Conflict of interest.
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Reprints and permissions
About this article
Meanwell, N.A. Reflections on a 40-year career in drug design and discovery. Med Chem Res 32 , 1208–1230 (2023). https://doi.org/10.1007/s00044-023-03070-6
Download citation
Accepted : 02 May 2023
Published : 03 July 2023
Issue Date : July 2023
DOI : https://doi.org/10.1007/s00044-023-03070-6
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Phosphodiesterase 3 inhibitors
- Prostacyclin agonists
- Maxi-K openers
- RSV inhibitors
- HCV NS3/4A, NS5A & NS5B inhibitors
- HIV-1 attachment & maturation inhibitors
- Find a journal
- Publish with us
- Track your research
COMMENTS
The department offers a PhD degree with no terminal MS option. In the first year, students typically conduct two to four 10-week rotations in different faculty labs to sample the breadth of research in the department. They then decide on a faculty mentor who will guide their dissertation research. Coursework requires students to become ...
The graduate program in Pharmaceutical Sciences spans the entire life cycle of a drug, from bench to bedside. The Medicinal Chemistry concentration focuses on drug discovery and development, part of the pre-clinical studies phase of the cycle. Pharmaceutical Sciences Ph.D. Programs Distinction through five interrelated training opportunities involving the entire life cycle of a drug.…
Coursework Once admitted to the PhD in Chemistry program, students are required to complete six graduate-level lecture courses during the first two years of full-time study. Of these courses, three must be one-semester introductory core courses selected from the four traditional areas of chemistry (CHE 501 and MCH 501 are required for the Medicinal Chemistry PhD), while the other three ...
Medicinal Chemistry and Drug Discovery. The PhD Program in Medicinal Chemistry educates and trains students in the design and synthesis of novel, biologically active compounds and in delineating their mechanisms of action using biochemical, biophysical, and pharmacological approaches. Research specializations are available in synthetic ...
Advanced entry into the Medicinal Chemistry and Drug Discovery PhD program requires a master's degree in pharmaceutical sciences or a related area and focuses on various advanced research courses and successful defense of the dissertation. An applicant's transcripts are required to be reviewed by the admissions committee to ensure they are eligible to be in the advanced entry program.
Design and synthesize novel biologically active compounds to address a broad range of social needs. The PhD Program in Medicinal Chemistry and Drug Discovery educates and trains students in the design and synthesis of novel, biologically active compounds and in delineating their mechanisms of action using biochemical, biophysical, and pharmacological approaches. Research specializations are ...
Our graduate program has been awarding PhD degrees to students for over seventy-five years and has a long and distinguished history of being among the top medicinal chemistry programs in the country. We are a diverse group of faculty, students, postdocs, researchers, and staff working at the interface of chemistry and biology. Research Interests.
A written statement explaining your interest in Medicinal Chemistry and why the graduate program at the University of Minnesota is a good fit for you. A brief summary of any prior work or research experience that has influenced your decision to pursue a PhD in Medicinal Chemistry with us. Your Resume or CV.
The KU Department of Medicinal Chemistry provides Ph.D. students a strong foundation in organic and medicinal chemistry with flexibility for additional emphasis in aspects of biochemistry, pharmacology and other biological sciences. Apply for Ph.D.
The Department of Medicinal Chemistry and Molecular Pharmacology (MCMP) is one of the top-rated programs in the country and is unique because it combines both medicinal chemistry and molecular pharmacology. Students in our PhD program will be trained in an environment that combines chemical and biological approaches, which is essential for ...
About the track. The medicinal chemistry track encompasses drug discovery and prepares you with the means to study the behavior of chemical substances at the molecular level. You will use computational, biochemical and cell-based screening technologies to identify natural and synthetic compounds with pharmacological activity.
Medicinal Chemistry (MS and PhD) Medicinal Chemistry is a multi-disciplinary field of study that utilizes synthetic organic chemistry in conjunction with developments in biochemistry, computational chemistry, molecular biology, and pharmacology to advance drug discovery. Faculty members within the Department of Medicinal Chemistry are actively ...
As a discipline, Medicinal Chemistry in the United States started with the appointment of Dr. F. F. Blicke as Assistant Professor of Pharmaceutical Chemistry in 1926. Prof. Blicke initiated the first graduate education program in Pharmaceutical Chemistry, focusing on synthetic organic chemistry.
Our master's and doctoral programs in Medicinal Chemistry focus on the theory and practice of drug design. Our strength — and what sets us apart from other programs — is our focus on chemistry and advanced biology. Not many universities emphasize both. Graduate students learn biological techniques to identify targets.
The Ph.D. in Medicinal Chemistry program requires a minimum of 60 post-baccalaureate semester hours: 48 credits of course work-including four (4) credits of seminar, and 12 credits of dissertation research. All Ph.D. programs at the Duquesne University Graduate School of Pharmaceutical Sciences in Pittsburgh, Pennsylvania are designed for ...
This program is STEM-designated, qualifying international students for an additional two years of OPT work in the United States. First-Year Experience (following completion of your MS degree) deepen your grasp of pharmaceutical science principles. study medicinal, organic, and bio-organic chemistry and spectroscopic analysis.
Department of Medicinal Chemistry. Ernest Mario School of Pharmacy. Rutgers, The State University of New Jersey. 160 Frelinghuysen Road. Piscataway, NJ 08854-8020. Phone: (848) 445-5381.
Located in the vibrant and multicultural city of Chicago, UIC's PhD Program in Pharmaceutical Sciences is one of the strongest and largest of its type in the United States. Our college is consistently ranked in the top ten in terms of funds secured annually from the National Institutes of Health and by US News and World Report.
PhD Program. Professor Wender discusses chemistry with his graduate students. Doctoral study in chemistry at Stanford University prepares students for research and teaching careers with diverse emphases in basic, life, medical, physical, energy, materials, and environmental sciences. The Department of Chemistry offers opportunities for graduate ...
Overview. The Master of Science in Medicinal Chemistry and Drug Discovery offered by the Department of Pharmaceutical Sciences, develops students' knowledge in the design, synthesis, and mechanisms of action of novel biologically active compounds using modern biochemical, biophysical, and pharmacological approaches.
The Medicinal Chemistry and Drug Discovery MS Program integrates aspects of contemporary medicinal chemistry and pharmacology, emphasizing topics most relevant to therapeutics design, discovery, and action. The core curriculum focuses on a combination of synthetic organic chemistry, bioorganic chemistry, analytical chemistry, and pharmacology ...
UNC Eshelman School of Pharmacy 301 Pharmacy Lane, CB#7355 Chapel Hill, NC 27599-7355 (919) 966-9786 Directions & Parking Chapel Hill Visitors Bureau
The minimum admission requirement for Ph.D. in Chemistry in USA is getting a bachelor's degree of 4 years with a GPA of 3.0 (83-86%). The GRE scores are not essential for getting admission, however, a good GRE/GMAT score can add value to the application. The top PhD universities in USA charges a tuition fees of 33-50 lakh INR .
The publication of this Perspective appeared to restore my standing with Derek Lowe, who commented on the article in his blog on March 22nd, 2011 (see Box 2), whilst also leading to invitations to present lectures on the topic at the annual "Drew Residential School on Medicinal Chemistry and Biology in Drug Discovery" and the biennial ...