
- DNA Replication
- Active Transport
- Cellular Receptors
- Endocytosis and Exocytosis
- Enzyme Inhibition
- Enzyme Kinetics
- Protein Structure
- Transcription of DNA
- Translation of DNA
- Anaerobic Respiration
- Electron Transport Chain
- Gluconeogenesis
- Calcium Regulation
- External Balance of Potassium
- Internal Balance of Potassium
- Sodium Regulation
- Cell Membrane
- Endoplasmic Reticulum
- Golgi Apparatus
- Mitochondria
- Blood Vessels
- Cellular Adaptations
- Epithelial Cells
- Muscle Histology
- Structure of Glands
- Control of Stroke Volume
- Control of Heart Rate
- Cardiac Cycle
- Cardiac Pacemaker Cells
- Conduction System
- Contraction of Cardiac Muscle
- Ventricular Action Potentials
- Blood Flow in Vessels
- Control of Blood Pressure
- Capillary Exchange
- Flow In Peripheral Circulation
- Venous Return
- Cardiac Muscle
- Hepatic Circulation
- Skeletal Muscle
- Airway Resistance
- Lung Volumes
- Mechanics of Breathing
- Gas Exchange
- Oxygen Transport in The Blood
- Transport of Carbon Dioxide in the Blood
- Ventilation-Perfusion Matching
- Chemoreceptors
- Cough Reflex
- Neural Control of Ventilation
- Respiratory Regulation of Acid-Base Balance
- Responses of The Respiratory System to Stress
- Regulation of Saliva
- Secretion of Saliva
- Gastric Acid Production
- Gastric Mucus Production
- Digestion and Absorption
- Histology and Cellular Function of the Small Intestine
- Absorption in the Large Intestine
- Large Intestinal Motility
- Bilirubin Metabolism
- Carbohydrate Metabolism in the Liver
- Lipid Metabolism in the Liver
- Protein and Ammonia Metabolism in the Liver
- Storage Functions of the Liver
- Bile Production
- Function of The Spleen
- Exocrine Pancreas
- Somatostatin
- Proximal Convoluted Tubule
- Loop of Henle
- Distal Convoluted Tubule and Collecting Duct
- Storage Phase of Micturition
- Voiding Phase of Micturition
- Antidiuretic Hormone
- Renin-Angiotensin-Aldosterone System
- Urinary Regulation of Acid-Base Balance
- Water Filtration and Reabsorption
- Development of the Reproductive System
- Gametogenesis
- Gonadotropins and the Hypothalamic Pituitary Axis
- Menstrual Cycle
- Placental Development
- Fetal Circulation
- Maternal Adaptations in Pregnancy
- Cells of the Nervous System
- Central Nervous System
- Cerebrospinal Fluid
- Neurotransmitters
- Peripheral Nervous System
- Action Potential
- Excitatory and Inhibitory Synaptic Signalling
- Resting Membrane Potential
- Synaptic Plasticity
- Synaptic Transmission
- Ascending Tracts
- Auditory Pathway
- Consciousness and Sleep
- Modalities of Sensation
- Pain Pathways
- Sensory Acuity
- Visual Pathway
- Descending Tracts
- Lower Motor Neurones
- Muscle Stretch Reflex
- Upper Motor Neurones
- Aqueous Humour
- Ocular Accommodation
- Thyroid Gland
- Parathyroid Glands
- Adrenal Medulla
- Zona Glomerulosa
- Zona Fasciculata
- Zona Reticularis
- Endocrine Pancreas
- The Hypothalamus
- Anterior Pituitary
- Posterior Pituitary
- White Blood Cells – Summary
- Barriers to Infection
- Infection Recognition Molecules
- Phagocytosis
- The Complement System

Antigen Processing and Presentation
- Primary and Secondary Immune Responses
- T Cell Memory
- Acute Inflammation
- Autoimmunity
- Chronic Inflammation
- Hypersensitivity Reactions
- Immunodeficiency
- Types of Immunity
- Antibiotics
- Viral Infection
- Blood Groups
- Coagulation
- Erythropoiesis
- Iron Metabolism
- Mononuclear Phagocyte System
Original Author(s): Antonia Round Last updated: 17th July 2023 Revisions: 9
- 1 Antigen Presentation
- 2.1 MHC Class I Molecules
- 2.2 MCH Class II Molecules
- 3.1 T Cell Receptors
- 3.2 Co-Receptors
- 4 Clinical Relevance – Autoimmune disease
T cells can only recognise antigens when they are displayed on cell surfaces. This is carried out by Antigen-presenting cells (APCs) , the most important of which are dendritic cells, B cells, and macrophages. APCs can digest proteins they encounter and display peptide fragments from them on their surfaces for other immune cells to recognise.
This process of antigen presentation allows T cells to “see” what proteins are present in the body and to form an adaptive immune response against them. In this article, we shall discuss antigen processing, presentation, and recognition by T cells.
Antigen Presentation
Antigens are delivered to the surface of APCs by Major Histocompatibility Complex (MHC) molecules. Different MHC molecules can bind different peptides. The MHC is highly polygenic and polymorphic which equips us to recognise a vast array of different antigens we might encounter. There are different classes of MHC, which have different functions:
- MHC class I molecules are found on all nucleated cells (not just professional APCs) and typically present intracellular antigens such as viruses.
- MHC class II molecules are only found on APCs and typically present extracellular antigens such as bacteria.
This is logical because should a virus be inside a cell of any type, the immune system needs to be able to respond to it. This also explains why pathogens inside human red blood cells (which are non-nucleated) can be difficult for the immune system to find, such as in malaria.
Whilst this is the general rule, in cross-presentation extracellular antigens can be presented by MHC class I, and in autophagy intracellular antigens can be presented by MHC class II.
Antigen Processing
Before an antigen can be presented, it must first be processed . Processing transforms proteins into antigenic peptides.
MHC Class I Molecules
Intracellular peptides for MHC class I presentation are made by proteases and the proteasome in the cytosol, then transported into the endoplasmic reticulum via TAP (Transporter associated with Antigen Processing) to be further processed.
They are then assembled together with MHC I molecules and travel to the cell surface ready for presentation.
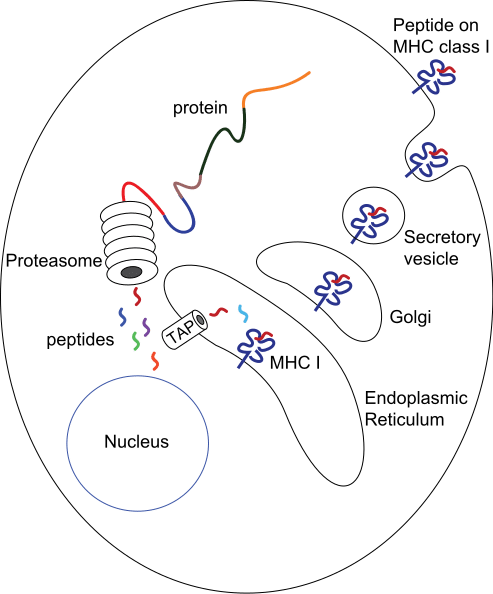
Fig 1 – Diagram demonstrating the production of peptides for MHC class I presentation
MCH Class II Molecules
The route of processing for exogenous antigens for MHC class II presentation begins with endocytosis of the antigen. Once inside the cell, they are encased within endosomes that acidify and activate proteases, to degrade the antigen.
MHC class II molecules are transported into endocytic vesicles where they bind peptide antigen and then travel to the cell surface.
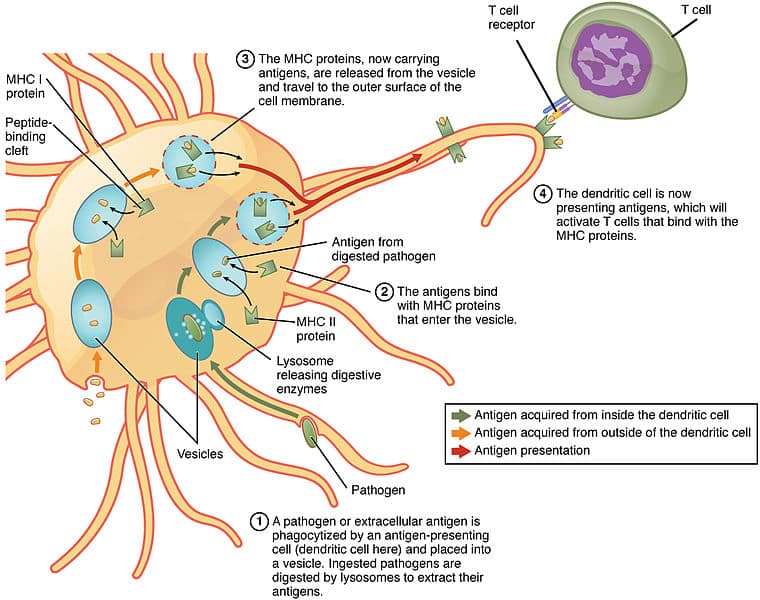
Fig 2 – Diagram showing processing of antigens for MHC Class II presentation by a dendritic cell
The antigen presented on MHCs is recognised by T cells using a T cell receptor (TCR) . These are antigen-specific .
T Cell Receptors
Each T cell has thousands of TCRs , each with a unique specificity that collectively allows our immune system to recognise a wide array of antigens.
This diversity in TCRs is achieved through a process called V(D)J recombination during development in the thymus. TCR chains have a variable region where gene segments are randomly rearranged, using the proteins RAG1 and RAG2 to initiate cleavage and non-homologous end joining to rejoin the chains.
The diversity of the TCRs can be further increased by inserting or deleting nucleotides at the junctions of gene segments; together forming the potential to create up to 10 15 unique TCRs.
TCRs are specific not only for a particular antigen but also for a specific MHC molecule. T cells will only recognise an antigen if a specific antigen with a specific MHC molecule is present: this phenomenon is called MHC restriction .
Co-Receptors
As well as the TCR, another T cell molecule is required for antigen recognition and is known as a co-receptor. These are either a CD4 or CD8 molecule:
- CD4 is present on T helper cells and only binds to antigen-MHC II complexes.
- CD8 is present on cytotoxic T cells and only binds to antigen-MHC I complexes.
This, therefore, leads to very different effects. Antigens presented with MHC II will activate T helper cells and antigens presented with MHC I activate cytotoxic T cells. Cytotoxic T cells will kill the cells that they recognise, whereas T helper cells have a broader range of effects on the presenting cell such as activation to produce antibodies (in the case of B cells) or activation of macrophages to kill their intracellular pathogens.
Clinical Relevance – Autoimmune disease
It is important to note that APCs may deliver foreign antigens or self-antigens. In the case of autoimmune diseases, self-antigens are presented to T cells, which then initiates an immune response against our own tissues.
For example, in Graves’ disease , TSHR (thyroid stimulating hormone receptor) acts as a self-antigen and is presented to T cells. This then activates B cells to produce autoantibodies against TSHRs in the thyroid. This results in the activation of TSHRs leading to hyperthyroidism and a possible goitre.
[start-clinical]
Clinical Relevance - Autoimmune disease
[end-clinical]
Found an error? Is our article missing some key information? Make the changes yourself here!
Once you've finished editing, click 'Submit for Review', and your changes will be reviewed by our team before publishing on the site.
We use cookies to improve your experience on our site and to show you relevant advertising. To find out more, read our privacy policy .
Privacy Overview
| Cookie | Duration | Description |
|---|---|---|
| cookielawinfo-checkbox-analytics | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics". |
| cookielawinfo-checkbox-functional | 11 months | The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional". |
| cookielawinfo-checkbox-necessary | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary". |
| cookielawinfo-checkbox-others | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other. |
| cookielawinfo-checkbox-performance | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance". |
| viewed_cookie_policy | 11 months | The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data. |
Antigen processing and presentation: Cytosolic and Endocytic pathway
August 3, 2020 Gaurab Karki Immunology 0

Antigen processing and Antigen presentation
- Antigen processing is a metabolic process that digests the proteins into peptides which can be displayed on the cell membrane together with a class-I or class-II MHC molecules and recognized by T-cells.
- Antigen presentation is the process by which certain cell in the body especially antigen presenting cells (APCs) express processed antigen on their cell surface along with MHC molecules in the form recognizable to T cell.
- If antigen is presented along with class-I MHC molecule, it is recognized by CD8 + Tc-cell and if presented along with class-II MHC molecule, it is recognized by CD4 + TH cells.
On the basis of types of antigen to be processed and presented, antigen processing and presenting pathway are of two types:
Cytosolic pathway of antigen processing and presentation
- Cytosolic pathway processed and presented the endogenous antigens i.e. those generated within cell eg. Viral infected cells, tumor cells and intracellular pathogens ( M . tuberculosis, Histoplasma capsulatum).
- The processed antigen is presented on the cell membrane with MHC-class I molecule which is recognized by CD8 + Tc-cell for degradation.
Steps involved in cytosolic pathways are:
- Proteolytic degradation of Ag (protein) into peptides
- Transportation of peptides from cytosol to RER
- Assembly of peptides with class I MHC molecules
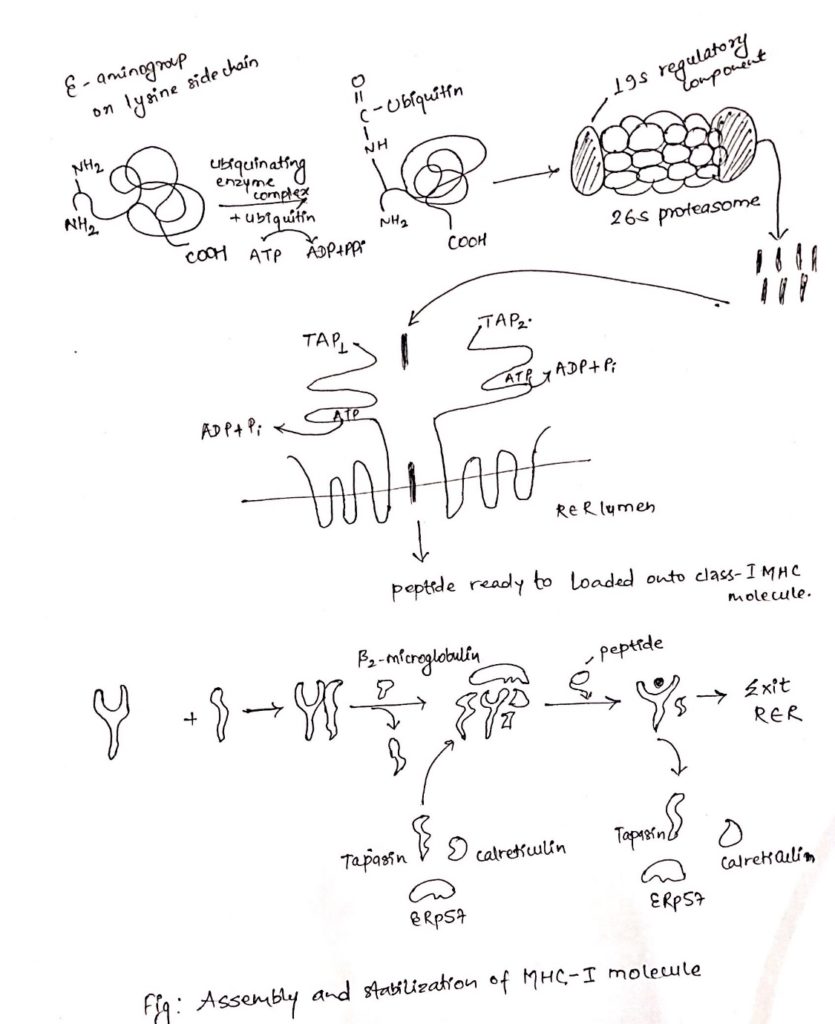
i. Proteolytic degradation of proteins into peptides:
- Intracellular proteineous antigen are larger in size to be bound to MHC molecule.
- So, it is degraded into short peptides of about 8-10 amino acids.
- These proteins are degraded by cytosolic proteolytic system present in cell called proteasome.
- The large (20S) proteasome is composed of 14 sub-units arranged in barrel-like structure of symmetrical rings.
- Some, but not all the sub-units have protease activity.
- Proteins enter the proteasome through narrow channel at each end.
- Many proteins targeted for proteolysis have a small protein called ubiquitin attached to them.
- Ubiquitin attached to them ubiquitin-protein complex consisting of 20S proteasome and 19S regulatory component added to it.
- The resulting 26S proteasome cleaves peptide bonds which is ATP-dependent process.
- Degradation of ubiquitin protein complex is thought to occur within the central hollow of the proteasome to release peptides.
ii. Transportation of peptides from cytosol to Rough Endoplasmic Reticulum (RER):
- Peptides generated in cytosol by proteasome are transported by TAP (transporter associated with antigen processing) into RER (Rough endoplasmic reticulum) by a process which require hydrolysis of ATP.
- TAP is membrane spanning heterodimer consisting of two proteins, TAP1 and TAP2.
- TAP has affinity for peptides having 8-16 amino acids.
- The optimal peptide length required by class-I MHC for binding is nine, which is achieved by trimming the peptides with the help of amino-peptidase present in RER. Eg. ERAP.
- In addition to it, TAP favor peptides with hydrophobic or basic carboxyl terminal amino acids, that preferred anchor residues for class-I MHC molecules.
- TAP deficiency can lead to a disease syndrome that has both immune-deficiency and auto-immunity aspects.
iii. Assembly of peptides with class-I MHC molecule:
- Like other proteins, the α-chain and β 2 microglobulin components of the class-I MHC molecule are synthesized on polysome along the rough endoplasmic reticulum.
- Assembly of these components into stable class-I MHC molecule that can exit the RRE require binding of peptides into peptide binding groove of class-I MHC molecules.
- The assembly process involves several steps and needs help of molecular chaperone.
- The first molecular chaperone involved in assembly of class-I MHC is calnexin.
- It is a resident membrane protein of RER.
- Calnexin associated with free class-I α-chain and promotes its folding.
- When β 2 -microglobulin binds class-I α-chain, calnexin is released and class-I MHC associates with another chaperone calreticulin and tapasin (TAP-associated protein).
- Tapasin brings TAP transporter carrying peptides to the proximity with class-I MHC molecule and allows to acquire the antigenic peptides.
- An additional protein with enzymatic activity, ERp57, form disulfide bond to tapasin and non-covalently associates with calreticulin to stabilize the interaction and allows release of MHC-I-class after acquiring antigenic peptides.
- As a consequence, the productive peptide binding with MHC of class-I releases from the complex of calreticulin, tapasin and ERp57, exit from RER and displays on the cell surface via golgi complex.
Endocytic pathway of antigen processing and presentation:
- The endocytic pathway processed and present the exogenous Ag. i.e. antigens generated outside the cells. E.g. Bacteria.
- At first APC phagocytosed, endocytosed or both, the antigen.
- Macrophage and dendritic cells internalize the antigen by both the process.
- While other APCs are non-phagocytic or poorly phagocytic. E.g. B cell internalize the antigen by receptor mediated endocytosis.
- Then antigen is processed and presented on the cell surface along with class-II MHC molecules which are recognized by CD4 + TH cell.
Steps involved in endocytic pathway:
- Peptide generation from internalized molecules (Ag) in endocytic vesicles.
- Transport of class-II MHC molecule to endocytic vesicles.
- Assembly of peptides with Class-II MHC molecules.
i. Peptide generation from internalized molecules (Ag) in endocytic vesicles:
- Once an antigen is internalized, it is degraded into peptides within compartments of endocytic processing pathway.
- The endocytic pathway appears to involve three increasingly acidic compartments, early endosomes (pH 6-6.5), late endosomes or endo-lysosome (pH 5-6) and lysosomes (pH 4.5-5).
- The internalized antigens move from early to late endosomes and finally to lysosomes, encountering hydrolytic enzymes and a lower pH in each compartment.
- Within the compartment, antigen is degraded into oligopeptides of about 13-18 residues.
- The mechanism by which internalized Ag moves from one endocytic compartment to next has not been clearly demonstrated.
- It has been suggested that early endosome move from periphery to inward to become late endosome and finally lysosomes.
- Alternatively, small transport vesicles may carry Ag from one compartment to next.
ii. Transport of class-II MHC molecule to endocytic vesicles:
- When class-II MHC molecules are synthesized within RER, three pairs of class-II αβ- chains associated with a pre-assembled trimer of a protein called invariant chain (Li, CD74).
- This trimeric protein prevents any endogenously antigen to bind to the cleft.
- The invariant chain consists of sorting signals in its cytoplasmic tail.
- It directs the transport of class-II MHC molecule to endocytic compartments from the trans-golgi network.
iii. Assembly of peptides with class-II MHC molecules:
- Class-II MHC-invariant chain complexes are transported from RER through golgi complex and golgi-network and through endocytic compartment, moving from early endosome to late endosome and finally to lysosome.
- The proteolytic activities increase in each compartment, so the invariant is slowly degraded.
- However, a short fragment of invariant chain remained termed as CLIP (Class-II associated invariant chain).
- CLIP physically occupies the peptide binding, cleft of class-II MHC molecule, presumably preventing any premature binding of antigenic peptides.
- A non-classical class-II MHC molecule known as HLA-DM is required to catalyze the exchange of CLIP with antigenic peptides.
- The reaction between HLA-DO, which binds to HLA-DM and lessens the efficiency of the exchange reactions.
- Conditions of higher acidity in endocytic compartment weakens the association of DM/DO and increase the possibility of antigenic peptide binding despite of DO.
- As with class-I MHC molecule, peptide binding is required to maintain the structure and stability of class-II MHC molecules.
- Once a peptide has bound the peptide-class II MHC complex is transported to the plasma membrane where neutral pH enables the complex to assume the compact and stable form.
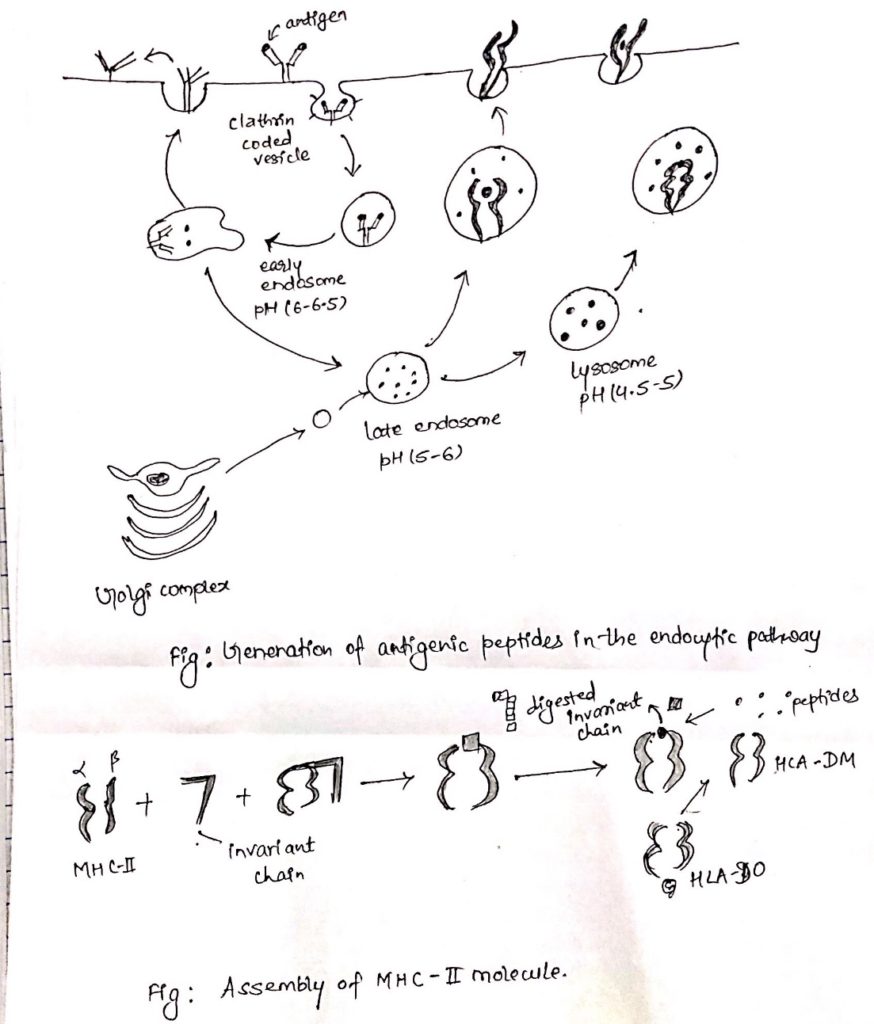
- Antigen processing and presentationCytosolic and Endocytic pathway
Copyright © 2024 | WordPress Theme by MH Themes
Select a Community
- MB 1 Preclinical Medical Students
- MB 2/3 Clinical Medical Students
- ORTHO Orthopaedic Surgery
Are you sure you want to trigger topic in your Anconeus AI algorithm?
You are done for today with this topic.
Would you like to start learning session with this topic items scheduled for future?
Antigen Processing and Presentation
- specialized antigen presenting cells (APCs) can activate the immune system
- killer T-cells can monitor the intracellular contents of all cells
- helper T-cells can be alerted to both intracellular and extracellular antigens
- endogenous antigens are proteins produced by the cell
- exogenous antigens are proteins that are taken up by the cell
- they can be stably exported to the cell surface
- they can be recognized specifically by T-cells
- recognize antigens loaded onto MHC class II
- recognize antigens loaded onto MHC class I
- degradation of proteins into component peptides
- translocation of the peptides into the appropriate compartment
- loading of the peptides onto MHC proteins
- the location from where the antigen originated
- the type of response that is required
- T-cell receptors can bind specifically to the peptide
- CD4 and CD8 can bind specifically to the corresponding MHC
- Integrins can bind APCs
- specific detection of antigens
- activation of T-cells
| Action | Numeric Key | Letter Key | Function Key |
|---|---|---|---|
| Show Bullets | S | Enter (frontside only) | |
| 20% | 1 | N | |
| 40% | 2 | H | |
| 60% | 3 | F | Enter (backside only) |
| 80% | 4 | E | |
| 100% | 5 | M | |
| Previous Card | Left Arrow | ||
| Next Card | N | Right Arrow | |
| Toss | 0 | T |
| Action | Numeric Key | Letter Key | Function Key |
|---|---|---|---|
| Choose 1 | 1 | ||
| Choose 2 | 2 | ||
| Choose 3 | 3 | ||
| Choose 4 | 4 | ||
| Choose 5 | 5 | ||
| Submit Response | Enter | ||
| Previous Question | Left Arrow | ||
| Next Question | N | Right Arrow | |
| Open/Close Bookmode | C | ||
| Open Image | Spacebar |
- - Antigen Processing and Presentation
Please Login to add comment

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- My Bibliography
- Collections
- Citation manager
Save citation to file
Email citation, add to collections.
- Create a new collection
- Add to an existing collection
Add to My Bibliography
Your saved search, create a file for external citation management software, your rss feed.
- Search in PubMed
- Search in NLM Catalog
- Add to Search
A guide to antigen processing and presentation
Affiliations.
- 1 Program in Cellular and Molecular Medicine, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA.
- 2 Society of Fellows, Harvard University, Cambridge, MA, USA.
- 3 Klarman Cell Observatory, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
- 4 Program in Cellular and Molecular Medicine, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA. [email protected].
- PMID: 35418563
- DOI: 10.1038/s41577-022-00707-2
Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high-affinity antibodies without T cell help. CD4 + T cells, which provide such help, use antigen-specific receptors that recognize major histocompatibility complex (MHC) molecules in complex with peptide cargo. Similarly, eradication of virus-infected cells often depends on cytotoxic CD8 + T cells, which rely on the recognition of peptide-MHC complexes for their action. The two major classes of glycoproteins entrusted with antigen presentation are the MHC class I and class II molecules, which present antigenic peptides to CD8 + T cells and CD4 + T cells, respectively. This Review describes the essentials of antigen processing and presentation. These pathways are divided into six discrete steps that allow a comparison of the various means by which antigens destined for presentation are acquired and how the source proteins for these antigens are tagged for degradation, destroyed and ultimately displayed as peptides in complex with MHC molecules for T cell recognition.
© 2022. Springer Nature Limited.
PubMed Disclaimer
Similar articles
- Invariant chain as a vehicle to load antigenic peptides on human MHC class I for cytotoxic T-cell activation. Wälchli S, Kumari S, Fallang LE, Sand KM, Yang W, Landsverk OJ, Bakke O, Olweus J, Gregers TF. Wälchli S, et al. Eur J Immunol. 2014 Mar;44(3):774-84. doi: 10.1002/eji.201343671. Epub 2013 Dec 27. Eur J Immunol. 2014. PMID: 24293164
- Mechanisms of antigen presentation. Jensen PE. Jensen PE. Clin Chem Lab Med. 1999 Mar;37(3):179-86. doi: 10.1515/CCLM.1999.034. Clin Chem Lab Med. 1999. PMID: 10353458 Review.
- Inhibition of the MHC class II antigen presentation pathway by human cytomegalovirus. Johnson DC, Hegde NR. Johnson DC, et al. Curr Top Microbiol Immunol. 2002;269:101-15. doi: 10.1007/978-3-642-59421-2_7. Curr Top Microbiol Immunol. 2002. PMID: 12224504 Review.
- Presentation of a self-peptide for in vivo tolerance induction of CD4+ T cells is governed by a processing factor that maps to the class II region of the major histocompatibility complex locus. Fedoseyeva EV, Tam RC, Orr PL, Garovoy MR, Benichou G. Fedoseyeva EV, et al. J Exp Med. 1995 Nov 1;182(5):1481-91. doi: 10.1084/jem.182.5.1481. J Exp Med. 1995. PMID: 7595218 Free PMC article.
- Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Münz C. Münz C. Trends Immunol. 2016 Nov;37(11):755-763. doi: 10.1016/j.it.2016.08.017. Epub 2016 Sep 22. Trends Immunol. 2016. PMID: 27667710 Review.
- The PDIA3-STAT3 protein complex regulates IBS formation and development via CTSS/MHC-II pathway-mediated intestinal inflammation. Weng C, Xu J, Ying X, Sun S, Hu Y, Wang X, He C, Lu B, Li M. Weng C, et al. Heliyon. 2024 Aug 28;10(17):e36357. doi: 10.1016/j.heliyon.2024.e36357. eCollection 2024 Sep 15. Heliyon. 2024. PMID: 39286134 Free PMC article.
- The emerging roles of UFMylation in the modulation of immune responses. Liang Z, Ning R, Wang Z, Kong X, Yan Y, Cai Y, He Z, Liu XG, Zou Y, Zhou J. Liang Z, et al. Clin Transl Med. 2024 Sep;14(9):e70019. doi: 10.1002/ctm2.70019. Clin Transl Med. 2024. PMID: 39259506 Free PMC article. Review.
- The contributions of T cell-mediated immunity to protection from vaccine-preventable diseases: A primer. Shapiro JR, Corrado M, Perry J, Watts TH, Bolotin S. Shapiro JR, et al. Hum Vaccin Immunother. 2024 Dec 31;20(1):2395679. doi: 10.1080/21645515.2024.2395679. Epub 2024 Aug 29. Hum Vaccin Immunother. 2024. PMID: 39205626 Free PMC article. Review.
- Revamping Hepatocellular Carcinoma Immunotherapy: The Advent of Microbial Neoantigen Vaccines. Liang J, Liao Y, Tu Z, Liu J. Liang J, et al. Vaccines (Basel). 2024 Aug 21;12(8):930. doi: 10.3390/vaccines12080930. Vaccines (Basel). 2024. PMID: 39204053 Free PMC article. Review.
- Chicken Secondary Lymphoid Tissues-Structure and Relevance in Immunological Research. Ceccopieri C, Madej JP. Ceccopieri C, et al. Animals (Basel). 2024 Aug 22;14(16):2439. doi: 10.3390/ani14162439. Animals (Basel). 2024. PMID: 39199973 Free PMC article. Review.
- Vyas, J. M., Van der Veen, A. G. & Ploegh, H. L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 8, 607–618 (2008). - PubMed - PMC
- Blees, A. et al. Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017). - PubMed
- Trowitzsch, S. & Tampe, R. Multifunctional chaperone and quality control complexes in adaptive immunity. Annu. Rev. Biophys. 49, 135–161 (2020). - PubMed
- Jensen, P. E. Recent advances in antigen processing and presentation. Nat. Immunol. 8, 1041–1048 (2007). - PubMed
- Call, M. E. & Wucherpfennig, K. W. The T cell receptor: critical role of the membrane environment in receptor assembly and function. Annu. Rev. Immunol. 23, 101–125 (2005). - PubMed
Publication types
- Search in MeSH
LinkOut - more resources
Full text sources.
- Nature Publishing Group
Other Literature Sources
- The Lens - Patent Citations
Research Materials
- NCI CPTC Antibody Characterization Program

- Citation Manager
NCBI Literature Resources
MeSH PMC Bookshelf Disclaimer
The PubMed wordmark and PubMed logo are registered trademarks of the U.S. Department of Health and Human Services (HHS). Unauthorized use of these marks is strictly prohibited.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 18 September 2024
Antigen presentation for central tolerance induction
- Ludger Klein 1 &
- Elisabetta Petrozziello 1
Nature Reviews Immunology ( 2024 ) Cite this article
836 Accesses
6 Altmetric
Metrics details
- Adaptive immunity
- Antigen-presenting cells
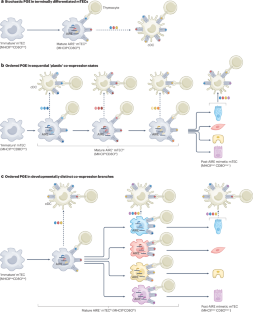
The extent of central T cell tolerance is determined by the diversity of self-antigens that developing thymocytes ‘see’ on thymic antigen-presenting cells (APCs). Here, focusing on insights from the past decade, we review the functional adaptations of medullary thymic epithelial cells, thymic dendritic cells and thymic B cells for the purpose of tolerance induction. Their distinct cellular characteristics range from unconventional phenomena, such as promiscuous gene expression or mimicry of peripheral cell types, to strategic positioning in distinct microenvironments and divergent propensities to preferentially access endogenous or exogenous antigen pools. We also discuss how ‘tonic’ inflammatory signals in the thymic microenvironment may extend the intrathymically visible ‘self’ to include autoantigens that are otherwise associated with highly immunogenic peripheral environments.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Factors that influence the thymic selection of CD8αα intraepithelial lymphocytes

B7-CD28 co-stimulation modulates central tolerance via thymic clonal deletion and Treg generation through distinct mechanisms

The emerging family of RORγt + antigen-presenting cells
Klein, L., Robey, E. A. & Hsieh, C. S. Central CD4 + T cell tolerance: deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 19 , 7–18 (2019).
Article CAS PubMed Google Scholar
Klein, L., Kyewski, B., Allen, P. M. & Hogquist, K. A. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol. 14 , 377–391 (2014).
Article CAS PubMed PubMed Central Google Scholar
Nedjic, J., Aichinger, M., Mizushima, N. & Klein, L. Macroautophagy, endogenous MHC II loading and T cell selection: the benefits of breaking the rules. Curr. Opin. Immunol. 21 , 92–97 (2009).
Anderson, M. S. et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298 , 1395–1401 (2002).
Kaiser, C., Bradu, A., Gamble, N., Caldwell, J. A. & Koh, A. S. AIRE in context: leveraging chromatin plasticity to trigger ectopic gene expression. Immunol. Rev. 305 , 59–76 (2022).
Sansom, S. N. et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24 , 1918–1931 (2014).
Meredith, M., Zemmour, D., Mathis, D. & Benoist, C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat. Immunol. 16 , 942–949 (2015).
Brennecke, P. et al. Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat. Immunol. 16 , 933–941 (2015). Together with Meredith et al. (2015), this study is the first to characterize gene expression in mTECs by single-cell transcriptomics, revealing that AIRE + mTECs segregate into subclusters of cells with stereotypic patterns of TRA co-expression.
Dhalla, F. et al. Biologically indeterminate yet ordered promiscuous gene expression in single medullary thymic epithelial cells. EMBO J. 39 , e101828 (2020).
Pinto, S. et al. Overlapping gene coexpression patterns in human medullary thymic epithelial cells generate self-antigen diversity. Proc. Natl Acad. Sci. USA 110 , E3497–E3505 (2013).
Gray, D., Abramson, J., Benoist, C. & Mathis, D. Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J. Exp. Med. 204 , 2521–2528 (2007).
Liiv, I. et al. AIRE-induced apoptosis is associated with nuclear translocation of stress sensor protein GAPDH. Biochem. Biophys. Res. Commun. 423 , 32–37 (2012).
Wang, X. et al. Post-Aire maturation of thymic medullary epithelial cells involves selective expression of keratinocyte-specific autoantigens. Front. Immunol. 3 , 19 (2012).
Metzger, T. C. et al. Lineage tracing and cell ablation identify a post-Aire-expressing thymic epithelial cell population. Cell Rep. 5 , 166–179 (2013).
Nishikawa, Y. et al. Temporal lineage tracing of Aire-expressing cells reveals a requirement for Aire in their maturation program. J. Immunol. 192 , 2585–2592 (2014).
Bornstein, C. et al. Single-cell mapping of the thymic stroma identifies IL-25-producing tuft epithelial cells. Nature 559 , 622–626 (2018).
Miller, C. N. et al. Thymic tuft cells promote an IL-4-enriched medulla and shape thymocyte development. Nature 559 , 627–631 (2018). Together with Bornstein et al. (2018), this study is the first to describe a population of mTECs with molecular and morphological characteristics of intestinal tuft cells.
Kadouri, N., Nevo, S., Goldfarb, Y. & Abramson, J. Thymic epithelial cell heterogeneity: TEC by TEC. Nat. Rev. Immunol. 20 , 239–253 (2020).
Givony, T. et al. Thymic mimetic cells function beyond self-tolerance. Nature 622 , 164–172 (2023). This study supports the idea that mimetic mTECs not only contribute to the induction of central tolerance but also regulate the homeostasis of other thymus-resident populations.
Michelson, D. A., Hase, K., Kaisho, T., Benoist, C. & Mathis, D. Thymic epithelial cells co-opt lineage-defining transcription factors to eliminate autoreactive T cells. Cell 185 , 2542–2558.e18 (2022). This study uses single-cell omics and histological analyses to reveal subsets of mTECs that express TRAs in a biologically logical manner, mirroring extra-thymic cell types while maintaining mTEC identity, that are hence referred to as mimetic cells.
Mathis, D. & Benoist, C. Aire. Annu. Rev. Immunol. 27 , 287–312 (2009).
Peterson, P., Org, T. & Rebane, A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat. Rev. Immunol. 8 , 948–957 (2008).
Lucas, B. et al. Diversity in medullary thymic epithelial cells controls the activity and availability of iNKT cells. Nat. Commun. 11 , 2198 (2020).
Guilliams, M. et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14 , 571–578 (2014).
Breed, E. R. et al. Type 2 cytokines in the thymus activate Sirpα + dendritic cells to promote clonal deletion. Nat. Immunol. 23 , 1042–1051 (2022). This paper shows that cytokine-induced activation of dendritic cells in the thymus substantially enforces central tolerance.
Ardouin, L. et al. Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity 45 , 305–318 (2016).
Voboril, M. et al. Toll-like receptor signaling in thymic epithelium controls monocyte-derived dendritic cell recruitment and T reg generation. Nat. Commun. 11 , 2361 (2020).
Atibalentja, D. F., Murphy, K. M. & Unanue, E. R. Functional redundancy between thymic CD8α + and Sirpα + conventional dendritic cells in presentation of blood-derived lysozyme by MHC class II proteins. J. Immunol. 186 , 1421–1431 (2011).
Baba, T., Nakamoto, Y. & Mukaida, N. Crucial contribution of thymic Sirpα + conventional dendritic cells to central tolerance against blood-borne antigens in a CCR2-dependent manner. J. Immunol. 183 , 3053–3063 (2009).
Kroger, C. J., Wang, B. & Tisch, R. Temporal increase in thymocyte negative selection parallels enhanced thymic SIRPα + DC function. Eur. J. Immunol. 46 , 2352–2362 (2016).
Raviola, E. & Karnovsky, M. J. Evidence for a blood–thymus barrier using electron-opaque tracers. J. Exp. Med. 136 , 466–498 (1972).
Vollmann, E. H. et al. Specialized transendothelial dendritic cells mediate thymic T-cell selection against blood-borne macromolecules. Nat. Commun. 12 , 6230 (2021). This study describes thymic dendritic cells that are positioned in immediate proximity to microvessels and extend cellular processes across the endothelial barrier, thereby sampling self-antigens from the bloodstream for central tolerance.
Barkauskas, D. S. et al. Extravascular CX3CR1 + cells extend intravascular dendritic processes into intact central nervous system vessel lumen. Microsc. Microanal. 19 , 778–790 (2013).
Guilliams, M., Lambrecht, B. N. & Hammad, H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol. 6 , 464–473 (2013).
Niess, J. H. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307 , 254–258 (2005).
Oh, J. et al. CD40 mediates maturation of thymic dendritic cells driven by self-reactive CD4 + thymocytes and supports development of natural regulatory T cells. J. Immunol. 200 , 1399–1412 (2018). This study shows that thymic dendritic cells mature and ‘optimize’ their tolerogenic potential in feedback response to cognate interactions with autoreactive CD4 + thymocytes.
Dudziak, D. et al. Differential antigen processing by dendritic cell subsets in vivo. Science 315 , 107–111 (2007).
Klein, L., Hinterberger, M., von Rohrscheidt, J. & Aichinger, M. Autonomous versus dendritic cell-dependent contributions of medullary thymic epithelial cells to central tolerance. Trends Immunol. 32 , 188–193 (2011).
Koble, C. & Kyewski, B. The thymic medulla: a unique microenvironment for intercellular self-antigen transfer. J. Exp. Med. 206 , 1505–1513 (2009).
Perry, J. S. A. et al. Transfer of cell-surface antigens by scavenger receptor CD36 promotes thymic regulatory T cell receptor repertoire development and allo-tolerance. Immunity 48 , 923–936.e4 (2018). This work proposes a mechanism for how mTEC-derived cell-surface antigen may be transferred to cDC1s to promote tolerance through indirect presentation.
Voboril, M. et al. A model of preferential pairing between epithelial and dendritic cells in thymic antigen transfer. eLife 11 , e71578 (2022).
Lei, Y. et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J. Exp. Med. 208 , 383–394 (2011).
Sornborger, A. et al. MiCASA is a new method for quantifying cellular organization. Nat. Commun. 8 , 15619 (2017).
Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8α + dendritic cells in cytotoxic T cell immunity. Science 322 , 1097–1100 (2008).
MacNabb, B. W. et al. Negligible role for deletion mediated by cDC1 in CD8 + T cell tolerance. J. Immunol. 202 , 2628–2635 (2019).
Leventhal, D. S. et al. Dendritic cells coordinate the development and homeostasis of organ-specific regulatory T cells. Immunity 44 , 847–859 (2016).
Perry, J. S. A. et al. Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity 41 , 414–426 (2014).
Lancaster, J. N. et al. Live-cell imaging reveals the relative contributions of antigen-presenting cell subsets to thymic central tolerance. Nat. Commun. 10 , 2220 (2019). This study uses sophisticated imaging methodology to simultaneously monitor thymocyte–APC interactions and intracellular signalling to dissect the relative contribution of mTECs versus dendritic cells to tolerogenic presentation of diverse mTEC-derived self-antigens.
Perera, J. et al. Self-antigen-driven thymic B cell class switching promotes T cell central tolerance. Cell Rep. 17 , 387–398 (2016).
Yamano, T. et al. Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity 42 , 1048–1061 (2015). This paper reveals a feed-forward loop that is driven by cognate interactions of thymic B cells with autoreactive CD4SP thymocytes and endows thymic B cells with potent tolerogenic features.
Kleindienst, P., Chretien, I., Winkler, T. & Brocker, T. Functional comparison of thymic B cells and dendritic cells in vivo. Blood 95 , 2610–2616 (2000).
Frommer, F. & Waisman, A. B cells participate in thymic negative selection of murine auto-reactive CD4 + T cells. PLoS ONE 5 , e15372 (2010).
Article PubMed PubMed Central Google Scholar
Lu, F. T. et al. Thymic B cells promote thymus-derived regulatory T cell development and proliferation. J. Autoimmun. 61 , 62–72 (2015).
Walters, S. N., Webster, K. E., Daley, S. & Grey, S. T. A role for intrathymic B cells in the generation of natural regulatory T cells. J. Immunol. 193 , 170–176 (2014).
Martinez, R. J. et al. Type III interferon drives thymic B cell activation and regulatory T cell generation. Proc. Natl Acad. Sci. USA 120 , e2220120120 (2023). This study shows that steady-state type III interferon signalling re-enforces thymic B cell licensing and thereby boosts the tolerogenic potential of thymic B cells.
Perera, J., Meng, L., Meng, F. & Huang, H. Autoreactive thymic B cells are efficient antigen-presenting cells of cognate self-antigens for T cell negative selection. Proc. Natl Acad. Sci. USA 110 , 17011–17016 (2013). Together with Perera et al. (2016), this study suggests that thymic B cells capture autoantigens through their BCR and present peptides derived from those autoantigens to developing thymocytes for negative selection.
Rother, M. B. et al. The human thymus is enriched for autoreactive B cells. J. Immunol. 197 , 441–448 (2016).
Baumgarth, N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 11 , 34–46 (2011).
Rudensky, A. Y., Mazel, S. M. & Yurin, V. L. Presentation of endogenous immunoglobulin determinant to immunoglobulin-recognizing T cell clones by the thymic cells. Eur. J. Immunol. 20 , 2235–2239 (1990).
Huszthy, P. C. et al. B cell receptor ligation induces display of V-region peptides on MHC class II molecules to T cells. Proc. Natl Acad. Sci. USA 116 , 25850–25859 (2019).
Lombard-Vadnais, F. et al. Activation-induced cytidine deaminase expression by thymic B cells promotes T-cell tolerance and limits autoimmunity. iScience 26 , 105852 (2023).
Dengjel, J. et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl Acad. Sci. USA 102 , 7922–7927 (2005).
Afzali, A. M. et al. B cells orchestrate tolerance to the neuromyelitis optica autoantigen AQP4. Nature 627 , 407–415 (2024). This paper shows that negative selection of AQP4-specific thymocytes in mice is largely dependent on the expression and presentation of AQP4 by thymic B cells, despite concomitant expression of AQP4 by mTECs.
Kwon, D. I. et al. Homeostatic serum IgE is secreted by plasma cells in the thymus and enhances mast cell survival. Nat. Commun. 13 , 1418 (2022).
Fonseca, V. R., Ribeiro, F. & Graca, L. T follicular regulatory (T FR ) cells: dissecting the complexity of T FR -cell compartments. Immunol. Rev. 288 , 112–127 (2019).
Spidale, N. A., Wang, B. & Tisch, R. Cutting edge: antigen-specific thymocyte feedback regulates homeostatic thymic conventional dendritic cell maturation. J. Immunol. 193 , 21–25 (2014).
Victora, G. D. & Nussenzweig, M. C. Germinal centers. Annu. Rev. Immunol. 40 , 413–442 (2022).
Laan, M. et al. Post-Aire medullary thymic epithelial cells and Hassall’s corpuscles as inducers of tonic pro-inflammatory microenvironment. Front. Immunol. 12 , 635569 (2021).
Martinez, R. J. & Hogquist, K. A. The role of interferon in the thymus. Curr. Opin. Immunol. 84 , 102389 (2023).
Ashby, K. M. et al. Sterile production of interferons in the thymus affects T cell repertoire selection. Sci. Immunol. 9 , eadp1139 (2024).
Article PubMed Google Scholar
Benhammadi, M. et al. IFN-λ enhances constitutive expression of MHC class I molecules on thymic epithelial cells. J. Immunol. 205 , 1268–1280 (2020).
Lee, Y. J., Holzapfel, K. L., Zhu, J., Jameson, S. C. & Hogquist, K. A. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat. Immunol. 14 , 1146–1154 (2013).
Lam, J. H., Smith, F. L. & Baumgarth, N. B cell activation and response regulation during viral infections. Viral Immunol. 33 , 294–306 (2020).
Lazear, H. M., Schoggins, J. W. & Diamond, M. S. Shared and distinct functions of type I and type III interferons. Immunity 50 , 907–923 (2019).
Pioli, K. T., Lau, K. H. & Pioli, P. D. Thymus antibody-secreting cells possess an interferon gene signature and are preferentially expanded in young female mice. iScience 26 , 106223 (2023).
Cordero, H. et al. Intrathymic differentiation of natural antibody-producing plasma cells in human neonates. Nat. Commun. 12 , 5761 (2021).
Derbinski, J., Schulte, A., Kyewski, B. & Klein, L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2 , 1032–1039 (2001).
Isaacson, P. G., Norton, A. J. & Addis, B. J. The human thymus contains a novel population of B lymphocytes. Lancet 2 , 1488–1491 (1987).
Leprince, C. et al. Thymic B cells from myasthenia gravis patients are activated B cells. Phenotypic Funct. Anal. J. Immunol. 145 , 2115–2122 (1990).
CAS Google Scholar
Vrolix, K. et al. Clonal heterogeneity of thymic B cells from early-onset myasthenia gravis patients with antibodies against the acetylcholine receptor. J. Autoimmun. 52 , 101–112 (2014).
Marx, A. et al. Thymus and autoimmunity. Semin. Immunopathol. 43 , 45–64 (2021).
Hidalgo, Y. et al. Thymic B cells promote germinal center-like structures and the expansion of follicular helper T cells in lupus-prone mice. Front. Immunol. 11 , 696 (2020).
Yasumizu, Y. et al. Myasthenia gravis-specific aberrant neuromuscular gene expression by medullary thymic epithelial cells in thymoma. Nat. Commun. 13 , 4230 (2022).
Srinivasan, J. et al. Age-related changes in thymic central tolerance. Front. Immunol. 12 , 676236 (2021).
Baran-Gale, J. et al. Ageing compromises mouse thymus function and remodels epithelial cell differentiation. eLife 9 , e56221 (2020).
Yang, S., Fujikado, N., Kolodin, D., Benoist, C. & Mathis, D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348 , 589–594 (2015). This work suggests that the intrathymic display of self-antigens is subject to age-dependent changes related to AIRE-independent differences in the processing and presentation of thymic stromal cell-derived peptides, resulting in ‘temporal windows’ of differential shaping of the T cell repertoire.
Stadinski, B. D. et al. A temporal thymic selection switch and ligand binding kinetics constrain neonatal Foxp3 + T reg cell development. Nat. Immunol. 20 , 1046–1058 (2019).
Griffith, A. V., Fallahi, M., Venables, T. & Petrie, H. T. Persistent degenerative changes in thymic organ function revealed by an inducible model of organ regrowth. Aging Cell 11 , 169–177 (2012).
Lancaster, J. N. et al. Central tolerance is impaired in the middle-aged thymic environment. Aging Cell 21 , e13624 (2022).
Cepeda, S. et al. Age-associated decline in thymic B cell expression of Aire and Aire-dependent self-antigens. Cell Rep. 22 , 1276–1287 (2018).
Velardi, E., Tsai, J. J. & van den Brink, M. R. M. T cell regeneration after immunological injury. Nat. Rev. Immunol. 21 , 277–291 (2021).
Bautista, J. L. et al. Single-cell transcriptional profiling of human thymic stroma uncovers novel cellular heterogeneity in the thymic medulla. Nat. Commun. 12 , 1096 (2021).
Park, J. E. et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 367 , eaay3224 (2020).
Bonasio, R. et al. Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat. Immunol. 7 , 1092–1100 (2006).
Zegarra-Ruiz, D. F. et al. Thymic development of gut-microbiota-specific T cells. Nature 594 , 413–417 (2021).
Yuseff, M. I., Pierobon, P., Reversat, A. & Lennon-Dumenil, A. M. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat. Rev. Immunol. 13 , 475–486 (2013).
Breed, E. R., Watanabe, M. & Hogquist, K. A. Measuring thymic clonal deletion at the population level. J. Immunol. 202 , 3226–3233 (2019).
Granados, D. P., Laumont, C. M., Thibault, P. & Perreault, C. The nature of self for T cells—a systems-level perspective. Curr. Opin. Immunol. 34 , 1–8 (2015).
Zerbino, D. R. et al. Ensembl 2018. Nucleic Acids Res. 46 , D754–D761 (2018).
Keane, P., Ceredig, R. & Seoighe, C. Promiscuous mRNA splicing under the control of AIRE in medullary thymic epithelial cells. Bioinformatics 31 , 986–990 (2015).
Danan-Gotthold, M., Guyon, C., Giraud, M., Levanon, E. Y. & Abramson, J. Extensive RNA editing and splicing increase immune self-representation diversity in medullary thymic epithelial cells. Genome Biol. 17 , 219 (2016).
Jansen, K. et al. RBFOX splicing factors contribute to a broad but selective recapitulation of peripheral tissue splicing patterns in the thymus. Genome Res. 31 , 2022–2034 (2021).
Padonou, F. et al. Aire-dependent transcripts escape Raver2-induced splice-event inclusion in the thymic epithelium. EMBO Rep. 23 , e53576 (2022).
Shilov, E. S., Gorshkova, E. A., Minnegalieva, A. R. & Potashnikova, D. M. Splicing pattern of mRNA in thymus epithelial cells limits the transcriptome available for negative selection of autoreactive T cells. Mol. Biol. 53 , 109–119 (2019).
Article CAS Google Scholar
Klein, L., Klugmann, M., Nave, K. A., Tuohy, V. K. & Kyewski, B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6 , 56–61 (2000).
Carter, J. A. et al. Transcriptomic diversity in human medullary thymic epithelial cells. Nat. Commun. 13 , 4296 (2022).
St-Pierre, C., Trofimov, A., Brochu, S., Lemieux, S. & Perreault, C. Differential features of AIRE-induced and AIRE-independent promiscuous gene expression in thymic epithelial cells. J. Immunol. 195 , 498–506 (2015).
Takaba, H. et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell 163 , 975–987 (2015).
Lammers, S. et al. Ehf and Fezf2 regulate late medullary thymic epithelial cell and thymic tuft cell development. Front. Immunol. 14 , 1277365 (2023).
Abramson, J., Giraud, M., Benoist, C. & Mathis, D. Aire’s partners in the molecular control of immunological tolerance. Cell 140 , 123–135 (2010).
Liiv, I. et al. DNA-PK contributes to the phosphorylation of AIRE: importance in transcriptional activity. Biochim. Biophys. Acta 1783 , 74–83 (2008).
Li, Y. et al. CCR4 and CCR7 differentially regulate thymocyte localization with distinct outcomes for central tolerance. eLife 12 , e80443 (2023).
Laan, M. et al. Autoimmune regulator deficiency results in decreased expression of CCR4 and CCR7 ligands and in delayed migration of CD4 + thymocytes. J. Immunol. 183 , 7682–7691 (2009).
Le Voyer, T. et al. Autoantibodies against type I IFNs in humans with alternative NF-κB pathway deficiency. Nature 623 , 803–813 (2023).
Meager, A. et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 3 , e289 (2006).
Meyer, S. et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell 166 , 582–595 (2016).
Morimoto, J. et al. Aire suppresses CTLA-4 expression from the thymic stroma to control autoimmunity. Cell Rep. 38 , 110384 (2022).
Michelson, D. A., Benoist, C. & Mathis, D. CTLA-4 on thymic epithelial cells complements Aire for T cell central tolerance. Proc. Natl Acad. Sci. USA 119 , e2215474119 (2022).
Taves, M. D., Donahue, K. M., Bian, J., Cam, M. C. & Ashwell, J. D. Aire drives steroid hormone biosynthesis by medullary thymic epithelial cells. Sci. Immunol. 8 , eabo7975 (2023).
Yano, M. et al. Aire controls the differentiation program of thymic epithelial cells in the medulla for the establishment of self-tolerance. J. Exp. Med. 205 , 2827–2838 (2008).
Anderson, M. S. et al. The cellular mechanism of Aire control of T cell tolerance. Immunity 23 , 227–239 (2005).
Hubert, F. X. et al. Aire regulates the transfer of antigen from mTECs to dendritic cells for induction of thymic tolerance. Blood 118 , 2462–2472 (2011).
Kuroda, N. et al. Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of Aire-deficient mice. J. Immunol. 174 , 1862–1870 (2005).
Hassall, A. H. Microscopic anatomy of the human body in health and disease (Highly, 1846).
Ragazzini, R. et al. Defining the identity and the niches of epithelial stem cells with highly pleiotropic multilineage potency in the human thymus. Dev. Cell 58 , 2428–2446.e9 (2023).
Michelson, D. A. & Mathis, D. Thymic mimetic cells: tolerogenic masqueraders. Trends Immunol. 43 , 782–791 (2022).
Consortium, U. I. G. et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 41 , 1330–1334 (2009).
Article Google Scholar
Liu, X. et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 42 , 658–660 (2010).
International Multiple Sclerosis Genetics Consortium et al. MANBA, CXCR5, SOX8, RPS6KB1 and ZBTB46 are genetic risk loci for multiple sclerosis. Brain 136 , 1778–1782 (2013).
Article PubMed Central Google Scholar
Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 19 , 539–549 (2019).
Surh, C. D. & Sprent, J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature 372 , 100–103 (1994).
Zhou, T. A. et al. Thymic macrophages consist of two populations with distinct localization and origin. eLife 11 , e75148 (2022).
Kroger, C. J., Spidale, N. A., Wang, B. & Tisch, R. Thymic dendritic cell subsets display distinct efficiencies and mechanisms of intercellular MHC transfer. J. Immunol. 198 , 249–256 (2017).
Schriek, P. & Villadangos, J. A. Trogocytosis and cross-dressing in antigen presentation. Curr. Opin. Immunol. 83 , 102331 (2023).
Skogberg, G., Telemo, E. & Ekwall, O. Exosomes in the thymus: antigen transfer and vesicles. Front. Immunol. 6 , 366 (2015).
Akiyama, T. et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity 29 , 423–437 (2008).
Hikosaka, Y. et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity 29 , 438–450 (2008).
Roberts, N. A. et al. Rank signaling links the development of invariant γδ T cell progenitors and Aire + medullary epithelium. Immunity 36 , 427–437 (2012).
Rossi, S. W. et al. RANK signals from CD4 + 3 – inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J. Exp. Med. 204 , 1267–1272 (2007).
White, A. J. et al. Sequential phases in the development of Aire-expressing medullary thymic epithelial cells involve distinct cellular input. Eur. J. Immunol. 38 , 942–947 (2008).
White, A. J. et al. An essential role for medullary thymic epithelial cells during the intrathymic development of invariant NKT cells. J. Immunol. 192 , 2659–2666 (2014).
Gao, H. et al. The lineage differentiation and dynamic heterogeneity of thymic epithelial cells during thymus organogenesis. Front. Immunol. 13 , 805451 (2022).
Irla, M. et al. Autoantigen-specific interactions with CD4 + thymocytes control mature medullary thymic epithelial cell cellularity. Immunity 29 , 451–463 (2008).
Download references
Acknowledgements
L.K. receives funding through the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under SFB 1054/3 Project A01 (210592381) and SFB-TRR 355/1 Project B01 (490846870). L.K. and E.P. receive DFG funding under Project number 456882036.
Author information
Authors and affiliations.
Institute for Immunology, Biomedical Center (BMC), Faculty of Medicine, LMU Munich, Planegg-Martinsried, Germany
Ludger Klein & Elisabetta Petrozziello
You can also search for this author in PubMed Google Scholar
Contributions
Both authors contributed equally to all aspects of the article.
Corresponding author
Correspondence to Ludger Klein .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Peer review
Peer review information.
Nature Reviews Immunology thanks Jakub Abramson, Lauren Ehrlich and the other anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
B cells that emerge early in life, are primarily found in the peritoneal cavity and have distinctive functions in innate immunity and the production of natural antibodies.
Conventional B cells found in secondary lymphoid organs, where they are crucial for adaptive immunity and antibody production.
Macroautophagy or microautophagy shuttles cytoplasmic material into lysosomal compartments and thereby intersects with the MHC class II loading pathway, so that peptides from endogenously expressed proteins can be loaded onto MHC class II.
A growth hormone predominantly secreted in the stomach, but also known to counteract age-dependent thymus involution.
Cells embedded in the epithelial lining of the intestine that endocytose luminal antigens and transport them to intraepithelial dendritic cells, macrophages and B cells.
The repertoires of peptides that are bound by MHC molecules.
(TRAs). An arbitrary term to describe genes with a confined expression pattern restricted to at most five peripheral cell types (in distinction from ubiquitously expressed self-antigens such as housekeeping genes).
Chemosensory and IL-25-expressing cells that are embedded in the epithelial lining of the intestine. Their name refers to characteristic brush-like microvilli.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Cite this article.
Klein, L., Petrozziello, E. Antigen presentation for central tolerance induction. Nat Rev Immunol (2024). https://doi.org/10.1038/s41577-024-01076-8
Download citation
Accepted : 25 July 2024
Published : 18 September 2024
DOI : https://doi.org/10.1038/s41577-024-01076-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

Warning: The NCBI web site requires JavaScript to function. more...
An official website of the United States government
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- Browse Titles
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.

Immunobiology: The Immune System in Health and Disease. 5th edition.
Chapter 5 antigen presentation to t lymphocytes.
In an adaptive immune response, antigen is recognized by two distinct sets of highly variable receptor molecules—the immunoglobulins that serve as antigen receptors on B cells and the antigen-specific receptors of T cells. As we saw in Chapter 3, T cells recognize only antigens that are displayed on cell surfaces. These antigens may derive from pathogens that replicate within cells, such as viruses or intracellular bacteria, or from pathogens or their products that cells internalize by endocytosis from the extracellular fluid. T cells can detect the presence of intracellular pathogens because infected cells display on their surface peptide fragments derived from the pathogens' proteins. These foreign peptides are delivered to the cell surface by specialized host-cell glycoproteins, the MHC molecules, which are also described in Chapter 3. The MHC glycoproteins are encoded in a large cluster of genes that were first identified by their potent effects on the immune response to transplanted tissues. For that reason, the gene complex was termed the major histocompatibility complex (MHC) . We now know that within this region of the genome, in addition to those genes encoding the MHC molecules themselves, are many genes whose products are involved in the production of the MHC:peptide complexes.
We will begin by discussing the mechanisms of antigen processing and presentation, whereby protein antigens are degraded into peptides inside cells and the peptides are then carried to the cell surface bound to MHC molecules. We will see that the two different classes of MHC molecule, known as MHC class I and MHC class II, deliver peptides from different cellular compartments to the surface of the infected cell. Peptides from the cytosol are bound to MHC class I molecules and are recognized by CD8 T cells, whereas peptides generated in vesicles are bound to MHC class II molecules and recognized by CD4 T cells. The two functional subsets of T cells are thereby activated to initiate the destruction of pathogens resident in these two different cellular compartments. Some CD4 T cells activate naive B cells that have internalized specific antigen, and thus also stimulate the production of antibodies to extracellular pathogens and their products.
In the second part of this chapter we will see that there are several genes for each class of MHC molecule: that is, the MHC is polygenic. Each of these genes has many variants: that is, the MHC is also highly polymorphic. Indeed, the most remarkable feature of the MHC class I and II genes is their genetic variability. MHC polymorphism has a profound effect on antigen recognition by T cells, and the combination of polygeny and polymorphism greatly extends the range of peptides that can be presented to T cells by each individual and each population at risk from an infectious pathogen.
- The generation of T-cell receptor ligands
- The major histocompatibility complex and its functions
- Summary to Chapter 5
- General references
- Section references
- Cite this Page Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001. Chapter 5, Antigen Presentation to T Lymphocytes.
Related Items in Bookshelf
- All Textbooks
Recent Activity
- Antigen Presentation to T Lymphocytes - Immunobiology Antigen Presentation to T Lymphocytes - Immunobiology
Your browsing activity is empty.
Activity recording is turned off.
Turn recording back on
Connect with NLM
National Library of Medicine 8600 Rockville Pike Bethesda, MD 20894
Web Policies FOIA HHS Vulnerability Disclosure
Help Accessibility Careers
- Environment
- Science & Technology
- Business & Industry
- Health & Public Welfare
- Topics (CFR Indexing Terms)
- Public Inspection
- Presidential Documents
- Document Search
- Advanced Document Search
- Public Inspection Search
- Reader Aids Home
- Office of the Federal Register Announcements
- Using FederalRegister.Gov
- Understanding the Federal Register
- Recent Site Updates
- Federal Register & CFR Statistics
- Videos & Tutorials
- Developer Resources
- Government Policy and OFR Procedures
- Congressional Review
- My Clipboard
- My Comments
- My Subscriptions
- Sign In / Sign Up
- Site Feedback
- Search the Federal Register
This site displays a prototype of a “Web 2.0” version of the daily Federal Register. It is not an official legal edition of the Federal Register, and does not replace the official print version or the official electronic version on GPO’s govinfo.gov.
The documents posted on this site are XML renditions of published Federal Register documents. Each document posted on the site includes a link to the corresponding official PDF file on govinfo.gov. This prototype edition of the daily Federal Register on FederalRegister.gov will remain an unofficial informational resource until the Administrative Committee of the Federal Register (ACFR) issues a regulation granting it official legal status. For complete information about, and access to, our official publications and services, go to About the Federal Register on NARA's archives.gov.
The OFR/GPO partnership is committed to presenting accurate and reliable regulatory information on FederalRegister.gov with the objective of establishing the XML-based Federal Register as an ACFR-sanctioned publication in the future. While every effort has been made to ensure that the material on FederalRegister.gov is accurately displayed, consistent with the official SGML-based PDF version on govinfo.gov, those relying on it for legal research should verify their results against an official edition of the Federal Register. Until the ACFR grants it official status, the XML rendition of the daily Federal Register on FederalRegister.gov does not provide legal notice to the public or judicial notice to the courts.
Proposed Rule
Design Updates: As part of our ongoing effort to make FederalRegister.gov more accessible and easier to use we've enlarged the space available to the document content and moved all document related data into the utility bar on the left of the document. Read more in our feature announcement .
Microbiology Devices; Reclassification of Antigen, Antibody, and Nucleic Acid-Based Hepatitis B Virus Assay Devices
A Proposed Rule by the Food and Drug Administration on 09/25/2024
This document has a comment period that ends in 59 days. (11/25/2024) Submit a public comment
Thank you for taking the time to create a comment. Your input is important.
Once you have filled in the required fields below you can preview and/or submit your comment to the Health and Human Services Department for review. All comments are considered public and will be posted online once the Health and Human Services Department has reviewed them.
You can view alternative ways to comment or you may also comment via Regulations.gov at https://www.regulations.gov/commenton/FDA-2024-N-3533-0001 .
- What is your comment about?
Note: You can attach your comment as a file and/or attach supporting documents to your comment. Attachment Requirements .
this will NOT be posted on regulations.gov
- Opt to receive email confirmation of submission and tracking number?
- Tell us about yourself! I am... *
- First Name *
- Last Name *
- State Alabama Alaska American Samoa Arizona Arkansas California Colorado Connecticut Delaware District of Columbia Florida Georgia Guam Hawaii Idaho Illinois Indiana Iowa Kansas Kentucky Louisiana Maine Maryland Massachusetts Michigan Minnesota Mississippi Missouri Montana Nebraska Nevada New Hampshire New Jersey New Mexico New York North Carolina North Dakota Ohio Oklahoma Oregon Pennsylvania Puerto Rico Rhode Island South Carolina South Dakota Tennessee Texas Utah Vermont Virgin Islands Virginia Washington West Virginia Wisconsin Wyoming
- Country Afghanistan Åland Islands Albania Algeria American Samoa Andorra Angola Anguilla Antarctica Antigua and Barbuda Argentina Armenia Aruba Australia Austria Azerbaijan Bahamas Bahrain Bangladesh Barbados Belarus Belgium Belize Benin Bermuda Bhutan Bolivia, Plurinational State of Bonaire, Sint Eustatius and Saba Bosnia and Herzegovina Botswana Bouvet Island Brazil British Indian Ocean Territory Brunei Darussalam Bulgaria Burkina Faso Burundi Cambodia Cameroon Canada Cape Verde Cayman Islands Central African Republic Chad Chile China Christmas Island Cocos (Keeling) Islands Colombia Comoros Congo Congo, the Democratic Republic of the Cook Islands Costa Rica Côte d'Ivoire Croatia Cuba Curaçao Cyprus Czech Republic Denmark Djibouti Dominica Dominican Republic Ecuador Egypt El Salvador Equatorial Guinea Eritrea Estonia Ethiopia Falkland Islands (Malvinas) Faroe Islands Fiji Finland France French Guiana French Polynesia French Southern Territories Gabon Gambia Georgia Germany Ghana Gibraltar Greece Greenland Grenada Guadeloupe Guam Guatemala Guernsey Guinea Guinea-Bissau Guyana Haiti Heard Island and McDonald Islands Holy See (Vatican City State) Honduras Hong Kong Hungary Iceland India Indonesia Iran, Islamic Republic of Iraq Ireland Isle of Man Israel Italy Jamaica Japan Jersey Jordan Kazakhstan Kenya Kiribati Korea, Democratic People's Republic of Korea, Republic of Kuwait Kyrgyzstan Lao People's Democratic Republic Latvia Lebanon Lesotho Liberia Libya Liechtenstein Lithuania Luxembourg Macao Macedonia, the Former Yugoslav Republic of Madagascar Malawi Malaysia Maldives Mali Malta Marshall Islands Martinique Mauritania Mauritius Mayotte Mexico Micronesia, Federated States of Moldova, Republic of Monaco Mongolia Montenegro Montserrat Morocco Mozambique Myanmar Namibia Nauru Nepal Netherlands New Caledonia New Zealand Nicaragua Niger Nigeria Niue Norfolk Island Northern Mariana Islands Norway Oman Pakistan Palau Palestine, State of Panama Papua New Guinea Paraguay Peru Philippines Pitcairn Poland Portugal Puerto Rico Qatar Réunion Romania Russian Federation Rwanda Saint Barthélemy Saint Helena, Ascension and Tristan da Cunha Saint Kitts and Nevis Saint Lucia Saint Martin (French part) Saint Pierre and Miquelon Saint Vincent and the Grenadines Samoa San Marino Sao Tome and Principe Saudi Arabia Senegal Serbia Seychelles Sierra Leone Singapore Sint Maarten (Dutch part) Slovakia Slovenia Solomon Islands Somalia South Africa South Georgia and the South Sandwich Islands South Sudan Spain Sri Lanka Sudan Suriname Svalbard and Jan Mayen Swaziland Sweden Switzerland Syrian Arab Republic Taiwan, Province of China Tajikistan Tanzania, United Republic of Thailand Timor-Leste Togo Tokelau Tonga Trinidad and Tobago Tunisia Turkey Turkmenistan Turks and Caicos Islands Tuvalu Uganda Ukraine United Arab Emirates United Kingdom United States United States Minor Outlying Islands Uruguay Uzbekistan Vanuatu Venezuela, Bolivarian Republic of Viet Nam Virgin Islands, British Virgin Islands, U.S. Wallis and Futuna Western Sahara Yemen Zambia Zimbabwe
- Organization Type * Company Organization Federal State Local Tribal Regional Foreign U.S. House of Representatives U.S. Senate
- Organization Name *
- You are filing a document into an official docket. Any personal information included in your comment text and/or uploaded attachment(s) may be publicly viewable on the web.
- I read and understand the statement above.
- Preview Comment
This document has been published in the Federal Register . Use the PDF linked in the document sidebar for the official electronic format.
- Document Details Published Content - Document Details Agencies Department of Health and Human Services Food and Drug Administration Agency/Docket Number Docket No. FDA-2024-N-3533 CFR 21 CFR 866 Document Citation 89 FR 78265 Document Number 2024-21932 Document Type Proposed Rule Pages 78265-78278 (14 pages) Publication Date 09/25/2024 Published Content - Document Details
- View printed version (PDF)
- Document Dates Published Content - Document Dates Comments Close 11/25/2024 Dates Text Either electronic or written comments on the proposed order must be submitted by November 25, 2024. Please see section X of this document for the proposed effective date when the new requirements apply and for the proposed effective date of a final order based on this proposed order. Published Content - Document Dates
This table of contents is a navigational tool, processed from the headings within the legal text of Federal Register documents. This repetition of headings to form internal navigation links has no substantive legal effect.
FOR FURTHER INFORMATION CONTACT:
Supplementary information:, i. background—regulatory authorities, ii. regulatory history of the devices, a. qualitative hbv antigen assays, b. hbv antibody assays (including qualitative and quantitative anti-hbs), c. quantitative hbv nucleic acid-based assays, iii. device description, iv. proposed reclassification and summary of reasons for reclassification, v. public health benefits and risks to health, vi. summary of data upon which the reclassification is based, vii. proposed special controls, viii. analysis of environmental impact, ix. paperwork reduction act of 1995, x. proposed effective date, xi. codification of orders, xii. references, list of subjects in 21 cfr part 866, part 866—immunology and microbiology devices.
Comments are being accepted - Submit a public comment .
FederalRegister.gov retrieves relevant information about this document from Regulations.gov to provide users with additional context. This information is not part of the official Federal Register document.
Microbiology Devices; Reclassification of Antigen, Antibody, and Nucleic Acid-Based Hepatitis B Virus Assay Device
- Sharing Enhanced Content - Sharing Shorter Document URL https://www.federalregister.gov/d/2024-21932 Email Email this document to a friend Enhanced Content - Sharing
- Print this document
Document page views are updated periodically throughout the day and are cumulative counts for this document. Counts are subject to sampling, reprocessing and revision (up or down) throughout the day.
This document is also available in the following formats:
More information and documentation can be found in our developer tools pages .
This PDF is the current document as it appeared on Public Inspection on 09/24/2024 at 8:45 am.
It was viewed 0 times while on Public Inspection.
If you are using public inspection listings for legal research, you should verify the contents of the documents against a final, official edition of the Federal Register. Only official editions of the Federal Register provide legal notice of publication to the public and judicial notice to the courts under 44 U.S.C. 1503 & 1507 . Learn more here .
Document headings vary by document type but may contain the following:
- the agency or agencies that issued and signed a document
- the number of the CFR title and the number of each part the document amends, proposes to amend, or is directly related to
- the agency docket number / agency internal file number
- the RIN which identifies each regulatory action listed in the Unified Agenda of Federal Regulatory and Deregulatory Actions
See the Document Drafting Handbook for more details.
Department of Health and Human Services
Food and drug administration.
- 21 CFR Part 866
- [Docket No. FDA-2024-N-3533]
Food and Drug Administration, HHS.
Proposed amendment; proposed order; request for comments.
The Food and Drug Administration (FDA, the Agency, or we) is proposing to reclassify qualitative hepatitis B virus (HBV) antigen assays, qualitative HBV antibody assays and quantitative assays that detect anti-HBs (antibodies to HBV surface antigen (HBsAg)), and quantitative HBV nucleic acid-based assays, all of which are postamendments class III devices, into class II (general controls and special controls), subject to premarket notification. FDA is also proposing three new device classification regulations along with the special controls that the Agency believes are necessary to provide a reasonable assurance of safety and effectiveness for each device.
Either electronic or written comments on the proposed order must be submitted by November 25, 2024. Please see section X of this document for the proposed effective date when the new requirements apply and for the proposed effective date of a final order based on this proposed order.
You may submit comments as follows. Please note that late, untimely filed comments will not be considered. The https://www.regulations.gov electronic filing system will accept comments until 11:59 p.m. Eastern Time at the end of November 25, 2024. Comments received by mail/hand delivery/courier (for written/paper submissions) will be considered timely if they are received on or before that date.
Electronic Submissions
Submit electronic comments in the following way:
- Federal Rulemaking Portal: https://www.regulations.gov . Follow the instructions for submitting comments. Comments submitted electronically, including attachments, to https://www.regulations.gov will be posted to the docket unchanged. Because your comment will be made public, you are solely responsible for ensuring that your comment does not include any confidential information that you or a third party may not wish to be posted, such as medical information, your or anyone else's Social Security number, or confidential business information, such as a manufacturing process. Please note that if you include your name, contact information, or other information that identifies you in the body of your comments, that information will be posted on https://www.regulations.gov .
- If you want to submit a comment with confidential information that you do not wish to be made available to the public, submit the comment as a written/paper submission and in the manner detailed (see “Written/Paper Submissions” and “Instructions”).
Written/Paper Submissions
Submit written/paper submissions as follows: ( print page 78266)
- Mail/Hand Delivery/Courier (for written/paper submissions): Dockets Management Staff (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
- For written/paper comments submitted to the Dockets Management Staff, FDA will post your comment, as well as any attachments, except for information submitted, marked and identified, as confidential, if submitted as detailed in “Instructions.”
Instructions: All submissions received must include the Docket No. FDA-2024-N-3533 for “Microbiology Devices; Reclassification of Antigen, Antibody, and Nucleic Acid-Based Hepatitis B Virus Assay Devices.” Received comments, those filed in a timely manner (see ADDRESSES ), will be placed in the docket and, except for those submitted as “Confidential Submissions,” publicly viewable at https://www.regulations.gov or at the Dockets Management Staff between 9 a.m. and 4 p.m., Monday through Friday Eastern Time, 240-402-7500.
- Confidential Submissions—To submit a comment with confidential information that you do not wish to be made publicly available, submit your comments only as a written/paper submission. You should submit two copies total. One copy will include the information you claim to be confidential with a heading or cover note that states “THIS DOCUMENT CONTAINS CONFIDENTIAL INFORMATION.” The Agency will review this copy, including the claimed confidential information, in its consideration of comments. The second copy, which will have the claimed confidential information redacted/blacked out, will be available for public viewing and posted on https://www.regulations.gov . Submit both copies to the Dockets Management Staff. If you do not wish your name and contact information to be made publicly available, you can provide this information on the cover sheet and not in the body of your comments and you must identify this information as “confidential.” Any information marked as “confidential” will not be disclosed except in accordance with 21 CFR 10.20 and other applicable disclosure law. For more information about FDA's posting of comments to public dockets, see 80 FR 56469 , September 18, 2015, or access the information at: https://www.govinfo.gov/content/pkg/FR-2015-09-18/pdf/2015-23389.pdf .
Docket: For access to the docket to read background documents, the plain language summary of the proposed order of not more than 100 words consistent with the “Providing Accountability Through Transparency Act,” or the electronic and written/paper comments received, go to https://www.regulations.gov and insert the docket number, found in brackets in the heading of this document, into the “Search” box and follow the prompts and/or go to the Dockets Management Staff, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852, 240-402-7500.
Maria Ines Garcia, Center for Devices and Radiological Health, Food and Drug Administration, 10903 New Hampshire Ave., Bldg. 66, Rm. 3104, Silver Spring, MD 20993, 301-796-7017, [email protected] .
The Federal Food, Drug, and Cosmetic Act (FD&C Act), as amended, establishes a comprehensive system for the regulation of medical devices intended for human use. Section 513 of the FD&C Act ( 21 U.S.C. 360c ) established three categories (classes) of devices, reflecting the regulatory controls needed to provide reasonable assurance of their safety and effectiveness. The three categories of devices are class I (general controls), class II (general controls and special controls), and class III (general controls and premarket approval).
Section 513(a)(1) of the FD&C Act defines the three classes of devices. Class I devices are those devices for which the general controls of the FD&C Act (controls authorized by or under sections 501, 502, 510, 516, 518, 519, or 520 ( 21 U.S.C. 351 , 352 , 360 , 360f , 360h , 360i , or 360j ) or any combination of such sections) are sufficient to provide reasonable assurance of safety and effectiveness; or those devices for which insufficient information exists to determine that general controls are sufficient to provide reasonable assurance of safety and effectiveness or to establish special controls to provide such assurance, but because the devices are not purported or represented to be for a use in supporting or sustaining human life or for a use which is of substantial importance in preventing impairment of human health, and do not present a potential unreasonable risk of illness or injury, are to be regulated by general controls (section 513(a)(1)(A) of the FD&C Act). Class II devices are those devices for which general controls by themselves are insufficient to provide reasonable assurance of safety and effectiveness, and for which there is sufficient information to establish special controls to provide such assurance, including the issue of performance standards, postmarket surveillance, patient registries, development and dissemination of guidelines, recommendations, and other appropriate actions the Agency deems necessary to provide such assurance (section 513(a)(1)(B) of the FD&C Act). Class III devices are those devices for which insufficient information exists to determine that general controls and special controls would provide a reasonable assurance of safety and effectiveness, and are purported or represented to be for a use in supporting or sustaining human life or for a use which is of substantial importance in preventing impairment of human health, or present a potential unreasonable risk of illness or injury (section 513(a)(1)(C) of the FD&C Act).
Devices that were not in commercial distribution before May 28, 1976 (generally referred to as “postamendments devices”) are automatically classified by section 513(f)(1) of the FD&C Act into class III without any FDA rulemaking process. Those devices remain in class III and require premarket approval, unless, and until: (1) FDA reclassifies the device into class I or II, or (2) FDA issues an order finding the device to be substantially equivalent, in accordance with section 513(i) of the FD&C Act, to a predicate device that does not require premarket approval. The Agency determines whether new devices are substantially equivalent to predicate devices by means of the premarket notification procedures in section 510(k) of the FD&C Act and part 807, subpart E ( 21 CFR part 807, subpart E ) of FDA's regulations.
A postamendments device that has been initially classified in class III under section 513(f)(1) of the FD&C Act may be reclassified into class I or class II under section 513(f)(3) of the FD&C Act. Section 513(f)(3) of the FD&C Act provides that FDA, acting by administrative order, can reclassify the device into class I or class II on its own initiative, or in response to a petition from the manufacturer or importer of the device. To change the classification of the device, the proposed new class must have sufficient regulatory controls to provide reasonable assurance of the safety and effectiveness of the device for its intended use.
FDA relies upon “valid scientific evidence”, as defined in section 513(a)(3) of the FD&C Act and 21 CFR 860.7(c)(2) , in the classification process to determine the level of regulation for devices. To be considered in the reclassification process, the “valid scientific evidence” upon which the Agency relies must be publicly available (see section 520(c) of the FD&C Act). ( print page 78267) Publicly available information excludes trade secret and/or confidential commercial information, e.g., the contents of a pending premarket approval application (PMA) (see section 520(c) of the FD&C Act).
In accordance with section 513(f)(3) of the FD&C Act, FDA is issuing this proposed order to reclassify qualitative HBV antigen assays intended for qualitative detection of HBV antigens as an aid in the diagnosis of acute or chronic HBV infection in specific populations, HBV antibody assays (including qualitative and quantitative anti-HBs) intended for use in the detection of antibodies to HBV, and quantitative HBV nucleic acid-based assays intended for use in the detection of HBV nucleic acid in specimens from individuals with antibody evidence of HBV infection, all of which are postamendments class III devices, into class II (general controls and special controls) subject to premarket notification, under three new device classification regulations with the names “Qualitative Hepatitis B Virus Antigen Assays,” “Hepatitis B Virus Antibody Assays,” and “Hepatitis B Virus Nucleic Acid-Based Assays.” FDA believes the standard in section 513(a)(1)(B) of the FD&C Act is met as there is sufficient information to establish special controls, which, in addition to general controls, will provide reasonable assurance of the safety and effectiveness of these devices. [ 1 ]
Section 510(m) of the FD&C Act provides that FDA may exempt a class II device from the premarket notification requirements under section 510(k) of the FD&C Act, if FDA determines that premarket notification is not necessary to provide reasonable assurance of the safety and effectiveness of the device. FDA has determined that premarket notification is necessary to provide a reasonable assurance of the safety and effectiveness of HBV antigen assays, HBV antibody assays, and HBV nucleic acid-based assays for their intended uses, therefore, the Agency does not intend to exempt these proposed class II devices from the requirement for premarket notification (510(k)) submission as provided under section 510(m) of the FD&C Act. If this proposed order is finalized, persons who intend to market this type of device must submit to FDA a premarket notification under section 510(k) of the FD&C Act prior to marketing the device.
Under section 513(f)(1) of the FD&C Act, qualitative HBV antigen assays, HBV antibody assays (including qualitative and quantitative anti-HBs), and quantitative HBV nucleic acid-based assays are automatically classified into class III because they were not introduced or delivered for introduction into interstate commerce for commercial distribution before May 28, 1976, and have not been found substantially equivalent to a device placed in commercial distribution after May 28, 1976, which was subsequently classified or reclassified into class II or class I. Therefore, they are subject to PMA requirements under section 515 of the FD&C Act ( 21 U.S.C. 360e ). Qualitative HBV antigen assays and HBV antibody assays (including qualitative and quantitative anti-HBs) are prescription devices and assigned product code LOM. Quantitative HBV nucleic acid-based assays are prescription devices and assigned product code MKT.
The first proposed device reclassification action applies to qualitative HBV antigen assay devices that are prescription in vitro diagnostic devices intended for qualitative detection of HBV antigens as an aid in the diagnosis of acute or chronic HBV infection in specific populations. On February 8, 2001, FDA approved its first HBV antigen assay (DiaSorin's ETI-EBK PLUS) for use in the qualitative detection of hepatitis Be antigen (HBeAg) in human serum or plasma (ethylenediaminetetraacetic acid (EDTA), citrate, or heparin) as indicative of a laboratory diagnosis of HBV infection through its PMA process under section 515 of the FD&C Act. On June 1, 2001, FDA approved its first HBV surface antigen (HBsAg) assay (Roche Elecsys HBsAg Immunoassay, Elecsys HBsAg Confirmatory, and Precicontrol HBsAg) for the qualitative detection of HBsAg in human serum or plasma (heparin, EDTA, sodium citrate) in adult pregnant and non-pregnant individuals. In a May 22, 2002, Federal Register notice ( 67 FR 36009 ), FDA announced the approval order and the availability of the Summary of Safety and Effectiveness Data (SSED) for these devices. Since the first approval order for an HBV antigen assay issued on February 8, 2001, FDA has approved 16 additional original PMAs for qualitative HBV antigen assays that are prescription devices intended for the detection of HBV antigens. These assays are intended as an aid in the diagnosis of acute or chronic HBV infection in conjunction with clinical findings and other diagnostic procedures ( e.g., HBV serology and antigen testing, liver function, etc.). These assays are not intended for use in screening of blood, plasma, cells, or tissue donors.
A review of the medical device reporting (MDR) databases indicates that there were 625 reported events for qualitative HBV antigen assays as of June 2024. Of these reported events, a significant majority of these were determined to be of no known impact or consequence to the patient. Events reported included false reactive results, false non-reactive results, incorrect or inadequate assay results, incorrect/inadequate/imprecise readings, improper or incorrect procedure or method, device operates differently than expected, and adverse event without identified device or use problems. Where incorrect results were obtained, it was not clear what the correct result should have been. As of June 2024, there have been no class III recalls, six class II recalls, and no class I recalls [ 2 ] involving qualitative HBV antigen assays. The class II recalls occurred since 2006 due to defective caps, device design, no marketing application, signal for reactive results, and biased results for biotin concentrations that were lower than indicated. No patient harm was identified. These facts, coupled with the low number of reported events that caused patient harm, indicate a good safety record for this device class. These recall events reflect the risks to health identified in section V below, and FDA believes the special controls proposed herein, in addition to general controls, can effectively mitigate the risks identified in these recalls.
The second type of devices this proposed reclassification order applies to are qualitative HBV antibody assays and quantitative anti-HBs assays that are prescription in vitro diagnostic devices intended for use in the detection of antibodies to HBV. These devices are intended to aid in the diagnosis of HBV infection in persons with signs and symptoms of hepatitis and in persons at risk for HBV infection. On September 29, 2000, FDA approved its first qualitative HBV antibody assay (Ortho-Clinical Diagnostics, Inc.'s Vitros ( print page 78268) Immunodiagnostic Products: Anti-HBS Reagent Pack/Anti-HBS Calibrators) for the qualitative in vitro determination of total antibody to hepatitis B surface antigen (anti-HBs) in human serum as an aid in determining susceptibility to HBV infection for individuals prior to or following HBV vaccination, or where vaccination status is unknown, and for use with other HBV serological markers for the laboratory diagnosis of HBV disease associated with HBV infection, through its PMA process under section 515 of the FD&C Act. In a March 12, 2001, Federal Register notice ( 66 FR 14390 ), FDA announced the approval order and the availability of the SSED for this device. On July 22, 2002, FDA approved its first quantitative Anti-HBs (Siemens Healthcare Diagnostics Products Ltd.'s Immulite 2000 XPI Anti-HBs) for the quantitative measurement of total antibodies to the hepatitis B surface antigen (anti-HBs) in human serum and plasma (heparinized or EDTA) as an aid in the determination of susceptibility to HBV infection for individuals prior to or following HBV vaccination, or where vaccination status is unknown, or for use with other HBV serological markers for the laboratory diagnosis of HBV disease associated with HBV infection, through its PMA process under section 515 of the FD&C Act.
Since the first approval order of a qualitative HBV antibody assay on September 29, 2000, FDA has approved 31 additional original PMAs for qualitative HBV antibody assays for the detection of antibodies to HBV. FDA has also approved six assays for quantitative anti-HBs detection. Qualitative HBV antibody assays and quantitative anti-HBs assays are intended to aid in the diagnosis of HBV infection in persons with signs and symptoms of hepatitis and in persons at risk for HBV infection in conjunction with clinical findings and other diagnostic procedures ( e.g., HBV serology and antigen testing, liver function, etc.). These assays are not intended for use in screening of blood, plasma, cells, or tissue donors.
A review of the MDR databases indicates that there were 1,107 reported events for HBV antibody assays between years 2001 and June 2024. Of these reported events, a significant majority of these were of no known impact to the patient, and only four resulted in impact to patients such as misdiagnosis or viral infection. Events reported included adverse events without identified device or use problem, disconnection/low assay results, false non-reactive results, false reactive results, false high assay results (for example, the first assay result had a low signal to cutoff (s/co) value and repeat testing produced a higher s/co value), incorrect assay results, inadequate assay results, and low assay results (for example, the first assay result was in the equivocal zone, repeat testing produced a non-reactive result, and testing with an alternate device produced a reactive result). In numerous cases, it was not possible to determine what the correct result should have been (further testing was not performed, insufficient sample volume, different assays were used). As of June 2024, FDA is aware of 4 class III recalls, 12 class II recalls, and no class I recalls for these devices. The class II recalls occurred in 2007, 2008, 2009, 2011, 2012, 2013, 2014, 2018, and 2019, and were related to issues such as false reactive results, false high assay results, defective caps, and errors in labeling, packaging, or software. No patient harm has been identified. These facts, coupled with the low number of reported events that impacted the patient, indicate a good safety record for this device class. These recall events reflect the risks to health identified in section V below, and FDA believes the special controls proposed herein, in addition to general controls, can effectively mitigate the risks identified in these recalls.
Finally, the third type of device this proposed reclassification order applies to are quantitative HBV nucleic acid-based assay devices for use as a prescription in vitro diagnostic device intended for use in the detection of HBV nucleic acid in specimens from individuals with antibody evidence of HBV infection. On September 4, 2008, FDA approved its first quantitative HBV nucleic assay (Roche Molecular Systems, Inc.'s COBAS TaqMan HBV Test For Use With The High Pure System), an in vitro nucleic acid amplification assay for the quantitation of HBV deoxyribonucleic acid (DNA) in human serum or plasma (EDTA) intended for use as an aid in the management of patients with chronic HBV infection undergoing antiviral therapy, through its PMA process under section 515 of the FD&C Act.
Since the first approval order, FDA has approved four additional original PMAs for quantitative HBV nucleic acid-based assays for the quantitative detection of HBV DNA. The detection of HBV DNA is used for management of patients undergoing antiviral therapy for assessing response to treatment and not as a diagnostic for HBV infection.
The following section provides examples of the different technologies used. The different technologies begin with specimen lysis and HBV DNA through hybridization with magnetic particles. The differences in the technologies occur with the method of amplification:
- In one technology, the target HBV DNA sequence is amplified. The presence of HBV amplification products is detected by measuring the fluorescence of the HBV probe that binds to the target. Similarly, the presence of the internal control amplification product is detected. In the absence of HBV or internal control target sequences, probe fluorescence is quenched. In the presence of HBV or internal control target, the HBV or internal control probes bind to their target.
- In another technology, target amplification occurs via transcription-based nucleic acid amplification by fluorescent labeled probes (torches). More torches hybridize when more amplicon is present creating a higher fluorescent signal. The time taken for the fluorescent signal to reach a threshold proportional to the starting HBV DNA concentration is measured in relation to internal controls.
A review of the MDR databases indicates that as of June 2024 there were 13 reported events for nucleic acid-based HBV DNA assays since the first reported event in 2009. MDRs were for the following reasons: (1) incorrect, inadequate, or imprecise result or readings; (2) high readings; and (3) non-reproducible results. Of these, two had no known impact or consequence to the patient and two occurred when the patient had no signs, symptoms, or conditions. As of June 2024, FDA is aware of one class III recall, five class II recalls, and no class I recalls for these devices. The class II recalls occurred between 2005 and 2022 and were related to issues such as misquantitation of high results for negative samples (carryover from a high positive sample tested adjacent to a negative sample may produce an incorrect positive result), liquid level detection of reagent cassette, under filled and over filled enzyme reagent vials in assay kits, software, and low level of recombinant HBV DNA found in one lot of reagent. These facts, coupled with the low number of reported events that impacted the patient, indicate a good safety record for this device class. These recall events reflect the risks to health identified in section V below, and FDA believes the special controls proposed herein, in addition to general controls, can effectively mitigate the risks identified in these recalls. ( print page 78269)
The HBV assays that are the subject of this proposed order are postamendments prescription in vitro diagnostic devices classified into class III under section 513(f)(1) of the FD&C Act.
A qualitative HBV antigen assay is a prescription in vitro diagnostic device intended for use in the qualitative detection of HBV antigens and for use as an aid in the diagnosis of HBV infection in specific populations. HBV antigen assays aid in the diagnosis of acute or chronic HBV infection. HBV antigen assays typically detect the presence of Hepatitis B surface antigen (HBsAg) or Hepatitis B e antigen (HBeAg). HBV antigens (HBsAg and HBeAg), when present in samples, bind to anti-HBs or anti-HBe antibodies to form a complex that is bound to a solid phase ( e.g., microparticles, microtiter plate or other technology). Detection of the complexes can be performed using different methods which measure the presence/absence of anti-HBs or anti-HBe antibodies in the sample.
Diagnosis of HBV infection should not be established based on a single assay result but should be determined in conjunction with clinical findings and other diagnostic procedures ( e.g., HBV serology and antigen testing, liver function, etc.). These assays are not intended for use in screening of blood, plasma, cells, or tissue donors.
A qualitative HBV antibody assay is a prescription in vitro diagnostic device intended for use in the qualitative detection of antibodies to HBV and for use as an aid in the diagnosis of HBV infection in specific populations. HBV antibody assays aid in the diagnosis of HBV infection in persons with signs and symptoms of hepatitis and in persons at risk for HBV infection. Antibody assays typically detect the presence of antibodies to HBsAg (anti-HBs), Hepatitis B core antigen (anti-HBc), or HBeAg (anti-HBe). Diagnosis of HBV infection should not be established based on a single assay result, but should be determined in conjunction with clinical findings and other diagnostic procedures ( e.g., HBV serology and antigen testing, liver function, etc.). These assays are not intended for use in screening of blood, plasma, cells, or tissue donors.
A quantitative assay that detects anti-HBs (antibodies to HBV surface antigen (HBsAg)) is a prescription in vitro diagnostic device that is intended for quantitative use to aid in the diagnosis of HBV infection in persons with signs and symptoms of hepatitis and in persons at risk for HBV infection. Detection of anti-HBs indicates a present or past infection with HBV and can be used in conjunction with clinical findings such as other HBV serological markers (detection of other HBV antigens and antibodies to HBV) for diagnosis of HBV infection. Anti-HBs assay results may be used as an aid in the determination of susceptibility to HBV infection in individuals prior to vaccination or when vaccination status is unknown.
In some device designs, HBV antibodies, when present in the sample, bind to HBV antigens to form a complex that is bound to a solid phase ( e.g., microparticles, microtiter plate, or other technology). Detection of complexes can be performed using different methods that measure the presence/absence of HBV antibodies in the sample.
A quantitative HBV nucleic acid-based assay is a prescription in vitro diagnostic device intended for use in the detection of HBV nucleic acid in specimens from individuals with antibody evidence of HBV infection. In these devices, the detection of HBV nucleic acid is used for management of patients undergoing antiviral therapy for assessing response to treatment and NOT as a diagnostic for HBV infection.
FDA is proposing to reclassify qualitative HBV antigen, HBV antibody assays (including qualitative and quantitative anti-HBs), and quantitative HBV nucleic acid-based assays from class III (general controls and premarket approval) to class II (general controls and special controls) and to establish new names for the device types that will be within the classification regulations. FDA proposes to revise 21 CFR part 866 to create three new device classification regulations with the names “Qualitative Hepatitis B Virus Antigen Assays,” “Hepatitis B Virus Antibody Assays,” and “Hepatitis B Virus Nucleic Acid-Based Assays.” FDA believes that these names and proposed identification language most accurately describe these devices.
- A Qualitative Hepatitis B Virus (HBV) Antigen Assay is tentatively identified as an in vitro diagnostic device intended for prescription use for qualitative use with human serum, plasma, or other matrices that aids in the diagnosis of chronic or acute HBV infection. HBV surface antigen (HBsAg) is also used for screening of HBV infection in pregnant women to identify neonates who are at risk of acquiring hepatitis B during perinatal period. The assay is not intended for screening of blood, plasma, cells, or tissue donors.
- A Hepatitis B Virus (HBV) Antibody Assay is tentatively identified as an in vitro diagnostic device intended for prescription use in the detection of antibodies to HBV in human serum and plasma, or other matrices, and or as an aid in the diagnosis of HBV infection in persons with signs and symptoms of hepatitis and in persons at risk for hepatitis B infection. In addition, anti-HBc IgM (IgM antibodies to core antigen) assay is indicative of recent HBV infection. Anti-HBs (antibodies to surface antigen) assay results may be used as an aid in the determination of susceptibility to HBV infection in individuals prior to or following HBV vaccination or when vaccination status is unknown. The assay is not intended for screening of blood, plasma, cells, or tissue donors. The assay is intended as an aid in diagnosis in conjunction with clinical findings and other diagnostic procedures.
- A Hepatitis B Virus (HBV) Nucleic Acid-Based Assay is tentatively identified as an in vitro diagnostic device intended for prescription use in the detection of HBV nucleic acid in specimens from individuals with antibody evidence of HBV infection. In these devices, the detection of HBV nucleic acid is used as an aid in the management of HBV-infected individuals. The assay is intended for use with human serum or plasma (and other matrices as applicable) from individuals with HBV. The assay is not intended for use as a donor screening assay for the presence of HBV nucleic acids in blood, blood products, plasma, cells, or tissue donors.
Based upon our review experience and consistent with the FD&C Act and FDA's regulations in 21 CFR 860.134 , FDA believes that these devices should be reclassified from class III into class II with special controls because there is sufficient information to establish special controls that, along with general controls, can provide reasonable assurance of the devices' safety and effectiveness.
FDA is proposing to reclassify the HBV assays that are the subject of this proposed order. On September 7, 2023, the Microbiology Devices Panel (Panel) of the Medical Devices Advisory Committee convened to discuss and make recommendations regarding the ( print page 78270) reclassification of HBV assays from class III (general controls and premarket approval) to class II (general controls and special controls) ( https://www.fda.gov/media/173609/download ). Panel members unanimously agreed that special controls, in addition to general controls, are necessary and sufficient to mitigate the risks to health of patients presented by these devices and to provide reasonable assurance of the safety and effectiveness of these devices (Refs. 1 and 2). The Panel agreed with FDA-identified risks and identified additional risk(s) and benefit(s) to include in the overall risk assessment. The Panel also discussed potential mitigation measure(s)/control(s) FDA should consider for each of the identified risks and recommended that, as part of any reclassification, the expected performance for these devices should remain the same. Notably, the performance of approved HBV antigen assays has generally been at least 97 percent sensitivity and 99 percent specificity. For approved anti-HBs, anti-Hbe, and anti-HBc total assays the sensitivity has generally been at least 95 percent, for approved anti-HBc IgM assays the sensitivity has been at least 86 percent, and for all HBV approved antibody assays the specificity has generally been above 97 percent.
FDA believes that at this time, sufficient data and information exist such that the risks identified in section V below can be mitigated by establishing special controls, and that these special controls, together with general controls, are necessary to provide a reasonable assurance of the safety and effectiveness of these HBV assays and therefore proposes these devices to be reclassified from class III (general controls and premarket approval) to class II (general controls and special controls). In accordance with section 513(f)(3) of the FD&C Act and 21 CFR part 860, subpart C , FDA is proposing to reclassify qualitative HBV antigen assays, HBV antibody assays (including qualitative and quantitative anti-HBs), and quantitative HBV nucleic acid-based assays from class III into class II, subject to premarket notification (510(k)) requirements. FDA believes that there is sufficient information available to FDA through FDA's accumulated experience with these devices from reviewing the PMAs for these HBV assays, and the Panel considerations and recommendations regarding the proposed special controls that FDA believes would effectively mitigate the risks to health identified in section V. Absent the special controls identified in this proposed order, general controls applicable to the devices are insufficient to provide reasonable assurance of the safety and effectiveness of the devices. FDA expects that the reclassification of these devices would enable more manufacturers to develop these assays such that patients would benefit from increased access to safe and effective tests.
FDA is proposing to create three separate classification regulations for HBV assays that will be reclassified from class III to class II. HBV assays are prescription in vitro diagnostic devices, and under this proposed order, if finalized, these devices will be identified as prescription in vitro diagnostic devices. As such, the devices must satisfy prescription labeling requirements for in vitro diagnostic products (see 21 CFR 809.10(a)(4) and (b)(5)(ii) ). In this proposed order, if finalized, FDA has identified the special controls under section 513(a)(1)(B) of the FD&C Act that, together with general controls, will provide a reasonable assurance of the safety and effectiveness of these assays.
FDA is also proposing to create a new product code for HBV antibody assays (including qualitative and quantitative anti-HBs) that will be assigned upon any finalization of this proposed order. Qualitative HBV antigen assays will continue to be assigned the product code LOM upon any finalization of this proposed order.
Section 510(m) of the FD&C Act provides that FDA may exempt a class II device from the premarket notification requirements under section 510(k) of the FD&C Act, if FDA determines that premarket notification is not necessary to provide reasonable assurance of the safety and effectiveness of the device. For these HBV assays, FDA has determined that premarket notification is necessary to provide a reasonable assurance of the safety and effectiveness of these devices. Therefore, the Agency does not intend to exempt these proposed class II devices from 510(k) requirements. If this proposed order is finalized, persons who intend to market a new HBV assay will no longer need to have a PMA for these devices but can instead submit to FDA a 510(k) and receive clearance prior to marketing the device. A 510(k) typically results in a shorter premarket review timeline compared to a PMA, which ultimately provides more timely access of these types of devices to patients.
FDA is providing a substantive summary of the valid scientific evidence concerning the public health benefits of the use of HBV assays (see also https://www.fda.gov/media/171770/download ), and the nature (and if known, the incidence) of the risks of the devices (see further discussion of the special controls being proposed to mitigate these risks in section VII of this proposed order).
HBV infection represents a significant global public health burden. According to the World Health Organization (WHO), in 2019 there were approximately 296 million people chronically infected people worldwide, with 1.5 million new HBV infections each year. [ 3 ] It is estimated by the Centers for Disease Control and Prevention (CDC) that chronic HBV infection in the United States affects at least between 580,000 to 1.17 million people with HBV infection in the United States; two-thirds of whom may be unaware of their infection. [ 4 ] HBV infection can be asymptomatic, and accordingly, many HBV-infected individuals are unaware of their HBV infection. Approximately 95 percent of adult patients with acute infection, defined as the first 6 months after infection, recover completely, and 5 percent of adults develop chronic HBV. [ 5 ] Infants born to women who are HbsAg-positive are at high risk of HBV infection. In absence of treatment, infants infected with HBV have a 90 percent risk of progression to chronic HBV and up to 25 percent of infants who acquire chronic HBV infection will die prematurely from HBV-related hepatocellular carcinoma or cirrhosis. [ 6 ] Patients who are tested and become aware that they are HBV infected may modify risk behaviors to prevent transmission to others and can be referred for treatment. Patients with chronic HBV infection have a risk of developing liver damage, liver cancer, or liver failure. They can also spread their infection to others. HBV can be reactivated in patients receiving immunosuppressive therapies, resulting in serious risk of liver failure or liver-associated death (Ref. 3). HBV is a vaccine-preventable liver infection.
With the initiation of the WHO Viral Hepatitis Elimination Plan [ 7 ] and the Department of Health and Human Services (HHS) Viral Hepatitis National ( print page 78271) Strategic Plan for the United States, [ 8 ] it is important for individuals to know their HBV infected status, to link HBV infected individuals to care, and to eliminate virus transmission. Therefore, diagnosis of patients with HBV infection through devices such as HBV antibody and antigen assays is essential to ensure that patients are linked to the appropriate care. Current CDC HBV Screening and Testing Recommendations include testing of the following groups: all adults 18 and older at least once in their lifetime using a triple panel test, pregnant women during pregnancy, people who are at ongoing risk for exposure, and anyone who requests HBV testing. [ 9 ]
FDA considered our accumulated experience with the regulation of these HBV assays, input from the Panel meeting, and postmarket information regarding these HBV assays, i.e., information from FDA's publicly available MDR, Manufacturer and User Facility Device Experience (MAUDE), and Medical Device Recall databases.
These HBV assays provide a benefit to the public health by informing individuals of their HBV infected status, linking HBV infected individuals to appropriate care, and aiding in eliminating virus transmission. Once an individual is tested and diagnosed as HBV infected, HBV nucleic acid testing is performed to inform treatment decisions. While HBV infection is treatable, it is not curable, which means that most people who start HBV antiviral treatment must continue it for life. The goal of current treatment is to suppress the virus and reduce the likelihood of long-term complications and transmission (Refs. 3 and 4). Thus, identifying individuals who are HBV infected, linking them to care, and managing their HBV infection to alleviate development of liver damage, liver cancer, liver failure, and potential HBV transmission would not only greatly impact public health but also go a long way towards helping the United States achieve HBV elimination.
Probable risks to health associated with the use of HBV assays include risks related to the risk of false results (false positives, false negatives, inaccurate low assay results, inaccurate high assay results, false reactive results, or false non-reactive results), failure to correctly interpret assay results, and failure to correctly operate the device. For HBV antigen and antibody assays, false positive results are generally referred to as false reactive results and false negative results are generally referred to as false non-reactive results. False results can lead to uninfected individuals receiving unnecessary further testing and treatment or infected individuals remaining undiagnosed and untreated. Undiagnosed and untreated individuals are likely to experience increases in morbidity and mortality and can spread the infection to others. FDA has identified the following additional specific risks to health associated with each of the HBV assays listed below.
Factors that may cause decreased assay sensitivity and/or an increased rate of false non-reactive results include, but are not limited to, the presence of interfering substances in the sample, acute infection at a stage that is too early for a device to detect the infection, and antigen concentrations that are too low to be detected by the device. Factors that may lead to false reactive results include device contamination from reactive samples, cross-reactivity with other antigens, or misinterpretation of invalid results as reactive.
- A false reactive assay result for HbeAg. Incorrectly interpreting the assay results as a reactive assay result or failing to correctly operate the assay causing a false reactive assay result may lead to continued treatment for hepatitis B with antiviral medication when it otherwise would not be indicated. Antiviral medication has risks including toxicity and more rarely allergic reactions. Over time, viral resistance in patients who are co-infected but undiagnosed with other viruses that are treated with the same antiviral medication, such as HIV, can lead to viral resistance.
- A false reactive assay result for HbsAg. Incorrectly interpreting the assay results as a reactive assay result or failing to correctly operate the assay causing a false reactive assay result may contribute to unnecessary additional testing, potentially delaying diagnosis of alternative causes of liver disease when present and may impact the psychological well-being of the patient. Factors that may increase the rate of false reactive assay reporting include cross-reactivity with antigens from other microorganisms or other disease conditions.
- A false non-reactive result for HbeAg. Incorrectly interpreting the assay results as a non-reactive assay result or failing to correctly operate the assay causing a false non-reactive assay result may lead to missing the opportunity for treatment of an HBV infected individual with antiviral medication or premature discontinuation of antiviral treatment when continuation of treatment is otherwise indicated should a clinician be falsely led to determine a patient has seroconverted HbeAg to anti-Hbe. Premature discontinuation of antiviral medication could result in adverse effects on patient health, such as cirrhosis, liver cancer, and liver damage, all of which are known to contribute to patient morbidity and mortality, or may contribute to public health risk by leading to virus transmission.
- A false non-reactive assay result for HbsAg. Incorrectly interpreting the assay results as a non-reactive assay result or failing to correctly operate the assay causing a false non-reactive assay result may delay or prevent a patient with HBV infection from being identified and linked to care. Missed identification of patients with chronic HBV infection could lead to adverse effects on patient health such as cirrhosis, liver cancer, and liver damage, all of which are known to contribute to patient morbidity and mortality. A false non-reactive HbsAg assay incorrectly interpreted as non-reactive also may contribute to public health risk by leading to virus transmission.
Factors that may cause decreased assay sensitivity and/or an increased rate of false non-reactive results include, but are not limited to, the presence of interfering substances in the sample, acute infection at a stage that is too early for a device to detect the infection, and antibody concentrations that are too low to be detected by the device. They also can be caused by misinterpretation of invalid results as non-reactive. Factors that may lead to false reactive results include device contamination from reactive samples, cross-reactivity with other antibodies, or misinterpretation of invalid results as reactive.
- A false reactive assay result for anti-HBs and anti-HBc. Incorrectly interpreting the assay results as a reactive assay result or failing to correctly operate the assay causing a false reactive assay result may lead to improper patient management. A false reactive antibody assay result could result in the unnecessary continuation of antiviral treatment. Antiviral medication has risks including toxicity and more rarely allergic reactions. Over time, viral resistance in patients who are co-infected but undiagnosed with other viruses that are treated with the same antiviral medication, such as HIV, can ( print page 78272) lead to viral resistance. Consequently, repeatedly false reactive results have the potential to lead to inappropriate patient management decisions.
- A false reactive assay result for anti-HBs. Incorrectly interpreting the assay results as a reactive assay result or failing to correctly operate the assay causing a false reactive assay result when the device is used as an aid in the determination of susceptibility to HBV infection in individuals prior to or following HBV vaccination or where vaccination status is unknown may cause a patient to be considered previously exposed and therefore immune to HBV or that the patient was successfully vaccinated. A false reactive result may cause the patient to not receive a vaccine, vaccine booster, hyperimmune globulin, and would be at higher risk of infection if exposed to HBV.
- A false reactive assay result for anti-Hbe. Incorrectly interpreting the assay results as a reactive assay result, or failing to correctly operate the assay causing a false reactive assay result may lead to missing the opportunity for treatment of HBV infection with antiviral medications in a subset of individuals for whom treatment would otherwise be indicated, or premature discontinuation of antiviral treatment when continuation of treatment is otherwise indicated should a clinician be falsely led to determine a patient has seroconverted HbeAg to anti-Hbe. Premature discontinuation of antiviral medication could result in adverse effects on patient health such as cirrhosis, liver cancer, and liver damage, all of which are known to contribute to patient morbidity and mortality, or may contribute to public health risk by leading to inadvertent transmission of virus by an infected individual.
- A false non-reactive assay result for anti-HBc. When the device is used as an aid in the diagnosis of HBV infection in patients with symptoms of hepatitis or who may be at risk for HBV infection, incorrectly interpreting the assay results as non-reactive assay result, or failing to correctly operate the assay causing a false non-reactive assay result may lead to non-diagnosis or a delay in diagnosis of HBV infection with an associated delay in therapy and potentially increased risk of HBV-related morbidity or mortality. Patients with active infection may unknowingly continue to infect others. False non-reactive results can also lead to unnecessary diagnostic evaluation if alternative etiologies of hepatitis are pursued. False non-reactive assay results may occur if the level of antibody in a specimen is below the limit of detection of the assay.
- A false non-reactive assay result for anti-HBs. When the device is used as an aid in the determination of susceptibility to HBV infection in individuals prior to or following HBV vaccination or where vaccination status is unknown, incorrectly interpreting the assay results as a non-reactive assay result or failing to correctly operate the assay causing a false non-reactive assay result may lead to unnecessary repeated vaccination for HBV.
- A false non-reactive assay result for anti-Hbe. Incorrectly interpreting the assay results as non-reactive assay result or failing to correctly operate the assay causing a false non-reactive assay result may lead to improper patient management, including continued treatment for HBV with antiviral medication. Antiviral medication has risks including toxicity and more rarely allergic reactions. Over time, viral resistance in patients who are co-infected but undiagnosed with other viruses using the same antiviral medication, such as HIV, can lead to viral resistance.
Decreased assay sensitivity and/or an increased rate of false negative assay reporting may occur with patient samples that contain different genotypes or rare de novo mutations in HBV genomic regions targeted by the device. In these situations, HBV viral load can transiently decrease and/or become undetectable in samples before the virus enters chronic replication.
- A false positive or falsely elevated quantitative HBV nucleic acid assay result. Incorrectly interpreting the assay results as a positive assay result or failing to correctly operate the assay causing a false positive assay result may negatively influence patient management decisions. Such decisions may include the administration or continuation of unnecessary antiviral treatment in patients with chronic HBV infection with its known toxicities and more rarely allergic reactions. Certain patients with falsely elevated HBV nucleic acid assay results may not undergo liver biopsy to investigate other causes of liver disease when the biopsy would otherwise be indicated for certain patients.
- A false negative or falsely decreased quantitative HBV nucleic acid assay result. Incorrectly interpreting the assay results as a negative assay result, or failing to correctly operate the assay causing a false negative assay result may negatively influence patient management decisions for patients with chronic HBV infection, including the withholding of treatment, failure to treat, or premature discontinuation of treating HBV infection when antiviral treatment is otherwise indicated or the choice of an inappropriate treatment. This could lead to adverse effects on patient health such as progressive liver disease, cirrhosis and/or hepatocellular carcinoma, and other cancers. Patients with active HBV replication also risk spreading the virus to others. Certain patients with falsely low HBV nucleic acid assay results may undergo liver biopsy to investigate other causes of liver disease.
The safety and effectiveness of these device types has become well established since the initial approval of the first qualitative HBV antibody assay in 2000, the first HBV antigen assay in 2001, and the first quantitative HBV nucleic acid-based assay in 2008. FDA has considered and analyzed the following information: (1) accumulated experience regulating these HBV assays, (2) input from the Panel meeting, and (3) postmarket information regarding HBV assays, i.e., information from FDA's publicly available MDR, MAUDE, and Medical Device Recall databases. The available evidence demonstrates that there are public health benefits derived from the use of HBV assays indicated for use to aid in diagnosis of HBV infection and/or for use to aid in the management of HBV infected patients, or as an aid in the determination of susceptibility to HBV infection (anti-HBs). In addition, the nature of the associated risks to health are known, and special controls can be established to sufficiently mitigate these risks.
Based on our review of the information described above, FDA has determined that special controls, in addition to general controls, are necessary to provide a reasonable assurance of safety and effectiveness for HBV assays, and that sufficient information exists to establish such special controls. Therefore, FDA, on its own initiative, is proposing to reclassify these postamendments devices from class III (general controls and premarket approval) into class II (general controls and special controls), subject to premarket notification (510(k)) requirements.
FDA believes that these devices can be classified into class II with the establishment of special controls. FDA believes that the following proposed special controls would mitigate each of ( print page 78273) the risks to health described in section V and that these special controls, in addition to general controls, would provide a reasonable assurance of safety and effectiveness for HBV assays. Tables 1 through 3 below demonstrate how FDA believes each risk to health described in section V would be mitigated by the proposed special controls for each device type.
The risk of inaccurate interpretation of assay results can be mitigated by special controls requiring certain labeling, including providing clearly stated warnings and limitations and information on principles of operation and procedures in performing the assay.
Risks associated with false results ( e.g., false non-reactive and false reactive assay results) and with the failure to correctly operate the device can be mitigated through a combination of special controls, including certain labeling requirements, certain design verification and validation information, and performance studies. Examples of verification and validation information to be included in the design of the device include documentation of performance specifications including analytical and clinical performance criteria. In addition, design verification and validation activities must include documentation of a complete device description, critical reagents, risk analysis strategies, lot release criteria, stability studies, and protocols. Required statements in labeling can aid in mitigating the failure of the device to perform as indicated, for example including a statement that use of the assay with specimen types other than those specifically identified for use with this device may cause inaccurate assay results. Special controls requiring additional labeling to provide a brief summary of the instructions for use can also mitigate these risks.
Table 1—Risks to Health and Mitigation Measures for Qualitative HBV Antigen Assays
The risk of falsely reactive, non-reactive, elevated, or lowered assay results can be mitigated by special controls requiring certain labeling, including providing clearly stated warnings and limitations and information on principles of operation and procedures in performing the assay.
Risks associated with the failure of the device to perform as indicated ( e.g., false non-reactive and false reactive assay results) can be mitigated through a combination of special controls, including certain labeling requirements, certain design verification and validation information, and performance studies. Examples of verification and validation information to be included in the design of the device include documentation of performance specifications including analytical and clinical performance criteria. In addition, design verification and validation activities must include documentation of a complete device description, critical reagents, risk analysis strategies, lot release criteria, stability studies, and protocols. Required statements in labeling can aid in mitigating the failure of the device to perform as indicated; for example, including a statement that use of the assay with specimen types other than those specifically identified for use with this device may cause inaccurate assay results.
Table 2—Risks to Health and Mitigation Measures for HBV Antibody Assays (Including Qualitative and Quantitative Anti-HBs)
The risk of falsely positive, negative, elevated, or lowered assay results can be mitigated by special controls requiring certain labeling, including providing clearly stated warnings and limitations, device description information, and detailed instructions in the device labeling regarding the interpretation of assay results and principles of operation and procedures in performing the assay.
Risks associated with the failure of the device to perform as indicated ( e.g., inaccurately low or high results, false negative results, and false positive assay results) can be mitigated through a combination of special controls related to certain labeling requirements, design verification and validation activities, and performance studies. Examples of verification and validation information to be included in the design of the device include documentation of a complete device description, calibrators, critical reagents, traceability, and lot release criteria. In addition, design verification and validation must include documentation of performance specifications, including analytical and clinical performance criteria. Required statements in labeling can aid in mitigating the occurrence of inaccurate results. The risks of false positive/false negative/falsely elevated/falsely lowered results due to decreased assay sensitivity can be mitigated by special controls related to certain labeling, design verification and validation activities, risk analysis strategies, and performance studies.
Table 3—Risks to Health and Mitigation Measures for Quantitative HBV Nucleic Acid-Based Assays
If this proposed order is finalized, qualitative HBV antigen assays, HBV antibody assays (including qualitative and quantitative anti-HBs), and quantitative HBV nucleic acid-based assays will be reclassified into class II (general controls and special controls) and would be subject to premarket notification requirements under section 510(k) of the FD&C Act. Firms submitting a 510(k) of the FD&C Act for such devices will be required to comply with the particular mitigation measures set forth in the special controls. FDA believes that adherence to the special controls, in addition to the general controls, is necessary to provide a reasonable assurance of safety and effectiveness of HBV assays.
We have determined under 21 CFR 25.34(b) that this action is of a type that does not individually or cumulatively have a significant effect on the human environment. Therefore, neither an environmental assessment nor an environmental impact statement is required.
While this proposed order contains no new collections of information, it does refer to previously approved FDA collections of information. The previously approved FDA collections of information are subject to review by the Office of Management and Budget (OMB) under the Paperwork Reduction Act of 1995 (PRA) ( 44 U.S.C. 3501-3521 ). The collections of information in 21 CFR part 820 have been approved under OMB control number 0910-0073; the collections of information in part 807, subpart E, have been approved under OMB control number 0910-0120; and the collections of information in 21 CFR parts 801 and 809 have been approved under OMB control number 0910-0485.
FDA proposes that any final order based on this proposed order become effective 30 days after the date of its publication in the Federal Register .
Under section 513(f)(3) of the FD&C Act, FDA may issue final orders to reclassify devices. FDA will continue to codify classifications and ( print page 78275) reclassifications in the Code of Federal Regulations (CFR). Changes resulting from final orders will appear in the CFR as newly codified orders. Therefore, under section 513(f)(3) of the FD&C Act, in the proposed order, we are proposing to codify qualitative hepatitis B virus antigen assays in the new § 866.3178, hepatitis B virus antibody assays (including qualitative and quantitative anti-HBs) in the new § 866.3179, and quantitative hepatitis B virus nucleic acid-based assays in the new § 866.3180, under which these HBV assays would be reclassified from class III into class II.
The following references marked with an asterisk (*) are on display at the Dockets Management Staff (see ADDRESSES ) and are available for viewing by interested persons between 9 a.m. and 4 p.m., Monday through Friday; they also are available electronically at https://www.regulations.gov . References without asterisks are not on public display at https://www.regulations.gov because they have copyright restriction. Some may be available at the website address, if listed. References without asterisks are available for viewing only at the Dockets Management Staff. Although FDA verified the website addresses in this document, please note that websites are subject to change over time.
*1. Summary Minutes Prepared for the September 7, 2023, Meeting of the Microbiology Devices Panel (available at https://www.fda.gov/media/173610/download ).
*2. Meeting Transcript Prepared for the September 7, 2023, Meeting of the Microbiology Devices Panel (available at https://www.fda.gov/media/173609/download ).
3. Terrault, N.A., A.S.F. Lok, B.J. McMahon, et al., “Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance.” Hepatology, 67(4): 1560-1599, 2018.
4. CDC, “Clinical Testing and Diagnosis for Hepatitis B,” https://www.cdc.gov/hepatitis-b/hcp/diagnosis-testing/index.html . Accessed July 11, 2024.
- Laboratories
- Medical devices
Therefore, under the Federal Food, Drug, and Cosmetic Act, and under authority delegated to the Commissioner of Food and Drugs, it is proposed that 21 CFR part 866 be amended as follows:
1. The authority citation for part 866 continues to read as follows:
Authority: 21 U.S.C. 351 , 360 , 360c , 360e , 360j , 360 l, 371.
2. Add § 866.3178 to subpart D to read as follows:
(a) Identification. A qualitative hepatitis B virus (HBV) antigen assay is identified as an in vitro diagnostic device intended for prescription use for qualitative use with human serum, plasma, or other matrices that aids in the diagnosis of chronic or acute HBV infection. HBV surface antigen (HbsAg) is also used for screening of HBV infection in pregnant women to identify neonates who are at risk of acquiring hepatitis B during perinatal period. The assay is not intended for screening of blood, plasma, cells, or tissue donors.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) The labeling required under § 809.10(b) of this chapter must include:
(i) A prominent statement that the assay is not intended for the screening of blood, plasma, cells, or tissue donors.
(ii) A detailed explanation of the principles of operation and procedures for performing the assay.
(iii) A detailed explanation of the interpretation of results.
(iv) Limitations, which must be updated to reflect current clinical practice and disease presentation and management. The limitations must include statements that indicate:
(A) The specimen types for which the device has been cleared, and that use of this assay with specimen types other than those specifically cleared for this device may result in inaccurate assay results.
(B) When appropriate, performance characteristics of the assay have not been established in populations of immunocompromised or immunosuppressed patients or other populations where assay performance may be affected.
(C) Diagnosis of hepatitis B infection should not be established on the basis of a single assay result but should be determined by a licensed healthcare professional in conjunction with the clinical presentation, history, and other diagnostic procedures.
(D) Detection of HBV antigens indicates a current infection with hepatitis B virus but does not differentiate between acute or chronic infection. False reactive HbsAg result may occur for up to 2 weeks after vaccination with HbsAg containing vaccine.
(E) Current methods for the detection of hepatitis B antigens may not detect all potentially infected individuals. A non-reactive assay result does not exclude the possibility of exposure to or infection with hepatitis B virus. A non-reactive assay result in individuals with prior exposure to hepatitis B may be due to but not limited to antigen levels below the detection limit of this assay or lack of antigen reactivity to the antibodies in this assay. HBV mutants lacking the ability to produce antigens have been reported. These may occur as “escape” mutants in the presence of anti-HBV antibodies and such patients may be infectious.
(F) Results obtained with this assay may not be used interchangeably with results obtained with a different manufacturer's assay.
(2) Design verification and validation must include the following:
(i) A detailed device description, including all parts that make up the device, ancillary reagents required but not provided, an explanation of the device methodology, design of the capture antibody(ies), external controls, and computational path from collected raw data to reported result ( e.g., how collected raw signals are converted into a reported signal and result), as applicable to the detection method and device design.
(ii) For devices with assay calibrators, the design and composition of all primary, secondary, and subsequent quantitation standards used for calibration as well as their traceability to a standardized reference material that FDA has determined is appropriate ( e.g., a recognized consensus standard). In addition, analytical testing must be performed following the release of a new lot of the standard material that was used for device clearance or approval, or when there is a transition to a new calibration standard.
(iii) Documentation and characterization ( e.g., supplier, determination of identity, purity, and stability) of all critical reagents (including description of the capture antibody(ies)), and protocols for maintaining product integrity throughout its labeled shelf life.
(iv) Risk analysis and management strategies, such as Failure Modes Effects Analysis and/or Hazard Analysis and Critical Control Points summaries and their impact on assay performance.
(v) Final release criteria to be used for manufactured assay lots with appropriate evidence that lots released ( print page 78276) at the extremes of the specifications will meet the identified analytical and clinical performance characteristics as well as stability.
(vi) Stability studies for reagents must include documentation of an assessment of real-time stability for multiple reagent lots using the indicated specimen types and must use acceptance criteria that ensure that analytical and clinical performance characteristics are met when stability is assigned based on the extremes of the acceptance range.
(vii) All stability protocols, including acceptance criteria.
(viii) Final release assay results for each lot used in clinical studies.
(ix) Reproducibility study data that includes the testing of three independent production lots.
(x) Detailed documentation of analytical performance studies conducted, as appropriate to the technology, specimen types tested, and intended use of the device, including, the limit of blank (LoB), limit of detection (LoD), cutoff, precision (reproducibility) including lot-to-lot and/or instrument-to-instrument precision, interference, cross reactivity, carryover, hook effect, seroconversion panel testing, matrix equivalency, prominent mutants/variants detection ( e.g., for HbsAg), specimen stability, reagent stability, and cross-genotype antigen detection sensitivity, when appropriate.
(xi) Analytical sensitivity of the assay is the same or better than that of other cleared or approved assays.
(xii) For devices with associated software or instrumentation, documentation must include a detailed description of device software, including software applications and hardware-based devices that incorporate software. The detailed description must include documentation of verification, validation, and hazard analysis and risk assessment activities, including an assessment of the impact of threats and vulnerabilities on device functionality and end users/patients as part of cybersecurity review.
(xiii) Detailed documentation and results from a clinical study. Performance must be analyzed relative to an FDA cleared or approved HBV antigen assay or a comparator that FDA has determined is appropriate. This study must be conducted using appropriate patient samples, with an appropriate number of HBV reactive and non-reactive samples in applicable risk and disease categories, and any applicable confirmatory testing. Additional relevant patient groups must be validated as appropriate. The samples must include prospective (sequential) samples for each identified specimen type and, as appropriate, additional characterized clinical samples. Samples must be sourced from geographically diverse areas. This study must be conducted in the appropriate settings by the intended users to demonstrate clinical performance.
3. Add § 866.3179 to subpart D to read as follows:
(a) Identification. A hepatitis B virus (HBV) antibody assay is identified as an in vitro diagnostic device intended for prescription use in the detection of antibodies to HBV in human serum, plasma, or other matrices, and as a device that aids in the diagnosis of HBV infection in persons with signs and symptoms of hepatitis and in persons at risk for hepatitis B infection. In addition, results from an anti-HBc IgM (IgM antibodies to core antigen) assay indicating the presence of anti-HBc IgM are indicative of recent HBV infection. Anti-HBs (antibodies to surface antigen) assay results may be used as an aid in the determination of susceptibility to HBV infection in individuals prior to or following HBV vaccination or when vaccination status is unknown. The assay is not intended for screening of blood, plasma, cells, or tissue donors. The assay is intended as an aid in diagnosis in conjunction with clinical findings and other diagnostic procedures.
(A) When appropriate, performance characteristics of the assay have not been established in populations of immunocompromised or immunosuppressed patients or other special populations where assay performance may be affected.
(B) Detection of HBV antibodies to a single viral antigen indicates a present or past infection with hepatitis B virus, but does not differentiate between acute, chronic, or resolved infection.
(C) The specimen types for which the device has been cleared, and that use of the assay with specimen types other than those specifically cleared for this device may result in inaccurate assay results.
(D) Diagnosis of hepatitis B infection should not be established on the basis of a single assay result but should be determined by a licensed healthcare professional in conjunction with the clinical presentation, history, and other diagnostic procedures.
(E) A non-reactive assay result may occur early during acute infection, prior to development of a host antibody response to infection, or when analyte levels are below the limit of detection of the assay.
(v) For devices intended for the quantitative detection of HBV antibodies (anti-HBs), in addition to the special controls listed in paragraphs (b)(1) and (2) of this section, labeling required under § 809.10(b) of this chapter must include:
(A) The assay calibrators' traceability to a standardized reference material that FDA has determined is appropriate ( e.g., a recognized consensus standard) and the limit of blank (LoB), limit of detection (LoD), limit of quantitation (LoQ), linearity, and precision to define the analytical measuring interval.
(B) Performance results of the analytical sensitivity study testing a standardized reference material that FDA has determined is appropriate ( e.g., a recognized consensus standard).
(i) Detailed device description, including all parts that make up the device, ancillary reagents required but not provided, an explanation of the device methodology, and design of the antigen(s) and capture antibody(ies) sequences, rationale for the selected epitope(s), degree of amino acid sequence conservation of the target, and the design and composition of all primary, secondary and subsequent standards used for calibration.
(ii) Documentation and characterization ( e.g., supplier, determination of identity, and stability) of all critical reagents (including description of the antigen(s) and capture antibody(ies)), and protocols for maintaining product integrity throughout its labeled shelf life.
(iii) Risk analysis and management strategies, such as Failure Modes Effects ( print page 78277) Analysis and/or Hazard Analysis and Critical Control Points summaries and their impact on assay performance.
(iv) Final release criteria to be used for manufactured assay lots with appropriate evidence that lots released at the extremes of the specifications will meet the identified analytical and clinical performance characteristics as well as stability.
(v) Stability studies for reagents must include documentation of an assessment of real-time stability for multiple reagent lots using the indicated specimen types and must use acceptance criteria that ensure that analytical and clinical performance characteristics are met when stability is assigned based on the extremes of the acceptance range.
(vi) All stability protocols, including acceptance criteria.
(vii) When applicable, analytical sensitivity of the assay is the same or better than that of other cleared or approved assays.
(viii) Analytical performance studies and results for determining the limit of blank (LoB), limit of detection (LoD), cutoff, precision (reproducibility), including lot-to-lot and/or instrument-to-instrument precision, interference, cross reactivity, carryover, hook effect, seroconversion panel testing, matrix equivalency, specimen stability, reagent stability, and cross-genotype antibody detection sensitivity, when appropriate.
(ix) For devices intended for the detection of antibodies for which a standardized reference material (that FDA has determined is appropriate) is available, the analytical sensitivity study and results testing the standardized reference material. Detailed documentation of that study and its results must be provided, including the study protocol, study report, testing results, and all statistical analyses.
(x) For devices with associated software or instrumentation, documentation must include a detailed description of device software, including software applications and hardware-based devices that incorporate software. The detailed description must include documentation of verification, validation, and hazard analysis and risk assessment activities, including an assessment of the impact of threats and vulnerabilities on device functionality and end users/patients as part of cybersecurity review.
(xi) Detailed documentation of clinical performance testing from a clinical study with an appropriate number of HBV reactive and non-reactive samples in applicable risk categories and conducted in the appropriate settings by the intended users. Performance must be analyzed relative to an FDA cleared or approved HBV antibody assay or a comparator that FDA has determined is appropriate. Additional relevant patient groups must be validated as appropriate. The samples must include prospective (sequential) samples for each identified specimen type and, as appropriate, additional characterized clinical samples. Samples must be sourced from geographically diverse areas.
(3) For any HBV antibody assay intended for quantitative detection of anti-HBV antibodies, the following special controls, in addition to those special controls listed in paragraphs (b)(1) and (2) of this section, also apply:
(i) Detailed documentation of the metrological calibration traceability hierarchy to a standardized reference material that FDA has determined is appropriate.
(ii) Detailed documentation of the following analytical performance studies conducted, as appropriate to the technology, specimen types tested, and intended use of the device, including upper and lower limits of quantitation (UloQ and LloQ, respectively), linearity using clinical samples, and an accuracy study using the recognized international standard material.
4. Add § 866.3180 to subpart D to read as follows:
(a) Identification. A nucleic acid-based hepatitis B virus (HBV) assay is identified as an in vitro diagnostic device intended for prescription use in the detection of HBV nucleic acid in specimens from individuals with antibody evidence of HBV infection. In these devices, the detection of HBV nucleic acid is used as an aid in the management of HBV-infected individuals. The assay is intended for use with human serum or plasma (and other matrices as applicable) from individuals with HBV. The assay is not intended for use as a donor screening assay for the presence of HBV nucleic acids in blood, blood products, plasma, cells, or tissue donors, or as a diagnostic assay to confirm the presence of HBV infection.
(1) Labeling required under § 809.10(b) of this chapter must include:
(i) A prominent statement that the assay is not intended for use as a screening assay for the presence of HBV DNA in blood or blood products, plasma, cells, or tissue donors, or as a diagnostic assay to confirm the presence of HBV infection.
(iv) Limitations, which must be updated to reflect current clinical practice and disease presentation and/or management. These limitations must include statements that indicate:
(A) Management of patients undergoing hepatitis B virus treatment should not be established on the basis of a single assay result but should be determined by a licensed healthcare professional in conjunction with the clinical presentation, history, and other diagnostic procedures, e.g., HBV serologic testing, liver function assays, liver elastography, etc.
(B) The specimen types for which the device has been cleared, and that use of this assay with specimen types other than those specifically cleared for this device may result in inaccurate assay results.
(C) The results obtained with this assay may not be used interchangeably with results obtained with a different manufacturer's assay.
(i) Detailed device description, including the device components, ancillary reagents required but not provided, and an explanation of the device methodology. Additional information appropriate to the technology must be included such as design of primers and probes, rationale for the selected gene targets, specifications for amplicon size, and degree of nucleic acid sequence conservation.
(iii) Documentation and characterization ( e.g., determination of the identity, supplier, purity, and stability) of all critical reagents (including nucleic acid sequences for primers and probes) and protocols for maintaining product integrity.
(iv) Risk analysis and management strategies demonstrating how risk ( print page 78278) control measures are implemented to address device system hazards, such as Failure Modes Effects Analysis and/or Hazard Analysis and Critical Control Points summaries and their impact on assay performance.
(v) Final release criteria to be used for manufactured assay lots with appropriate evidence that lots released at the extremes of the specification will meet the identified analytical and clinical performance characteristics as well as stability.
(viii) Detailed documentation of analytical performance studies conducted as appropriate to the technology, specimen types tested, and intended use of the device, including limit of detection (LoD), linearity, precision, endogenous and exogenous interferences, cross-reactivity, carryover, matrix equivalency, sample and reagents stability, and as applicable, upper and lower limits of quantitation (ULoQ and LLoQ, respectively). Samples selected for use must be from subjects with clinically relevant circulating genotypes in the United States. Cross-reactivity studies must include samples from HBV nucleic acid negative subjects with other viral or non-viral causes of liver disease, including autoimmune hepatitis, alcoholic liver disease, chronic hepatitis C virus (HCV), primary biliary cirrhosis, and nonalcoholic steatohepatitis, when applicable. The effect of each identified nucleic-acid isolation and purification procedure on detection must be evaluated.
(ix) For devices with associated software or instrumentation, documentation must include a detailed description of device software, including software applications and hardware-based devices that incorporate software. The detailed description must include documentation of verification, validation, and hazard analysis and risk assessment activities, including an assessment of the impact of threats and vulnerabilities on device functionality and end users/patients as part of cybersecurity review.
(x) Detailed documentation of performance from a clinical study with a design and number of clinical samples (appropriately statistically powered) that is appropriate for the intended use of the device as well as conducted in the appropriate settings by the intended users. The samples must include prospective (sequential) samples for each claimed specimen type and, as appropriate, additional characterized clinical samples. Samples must be sourced from geographically diverse areas.
Dated: September 20, 2024.
Lauren K. Roth,
Associate Commissioner for Policy.
1. FDA notes that the “ACTION” caption for this proposed order is styled as “Proposed amendment; proposed order,” rather than “Proposed order.” Beginning in December 2019, this editorial change was made to indicate that the document “amends” the Code of Federal Regulations. The change was made in accordance with the Office of the Federal Register's (OFR) interpretations of the Federal Register Act ( 44 U.S.C. chapter 15 ), its implementing regulations ( 1 CFR 5.9 and parts 21 and 22 ), and the Document Drafting Handbook.
2. Class I, II, and III recalls are defined in 21 CFR 7.3(m) .
3. https://www.who.int/news-room/fact-sheets/detail/hepatitis-b . Accessed on July 12, 2024.
4. Centers for Disease Control and Prevention—Clinical Overview of Hepatitis B (Available at https://www.cdc.gov/hepatitis-b/hcp/clinical-overview/index.html ). Accessed on July 12, 2024.
5. Ibid.
6. Ibid.
7. https://www.who.int/health-topics/hepatitis/elimination-of-hepatitis-by-2030#tab=tab_1 . Accessed on July 12, 2024.
8. https://www.hhs.gov/sites/default/files/Viral-Hepatitis-National-Strategic-Plan-2021-2025.pdf . Accessed on July 12, 2024.
9. https://www.cdc.gov/hepatitis/hbv/index.htm . Accessed on July 12, 2024.
[ FR Doc. 2024-21932 Filed 9-24-24; 8:45 am]
BILLING CODE 4164-01-P
- Executive Orders
Reader Aids
Information.
- About This Site
- Legal Status
- Accessibility
- No Fear Act
- Continuity Information
IMAGES
COMMENTS
Antigen Processing and Presentation. In order to be capable of engaging the key elements of adaptive immunity (specificity, memory, diversity, self/nonself discrimination), antigens have to be processed and presented to immune cells. Antigen presentation is mediated by MHC class I molecules, and the class II molecules found on the surface of ...
This process of antigen presentation allows T cells to "see" what proteins are present in the body and to form an adaptive immune response against them. In this article, we shall discuss antigen processing, presentation, and recognition by T cells. ... Antigen Presentation. The antigen presented on MHCs is recognised by T cells using a T ...
Abstract. Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high-affinity antibodies without T cell help. CD4 + T cells, which provide such ...
Antigen presentation is a vital immune process that is essential for T cell immune response triggering. Because T cells recognize only fragmented antigens displayed on cell surfaces, antigen processing must occur before the antigen fragment can be recognized by a T-cell receptor. Specifically, the fragment, bound to the major histocompatibility ...
Antigen processing and Antigen presentation. Antigen processing is a metabolic process that digests the proteins into peptides which can be displayed on the cell membrane together with a class-I or class-II MHC molecules and recognized by T-cells. Antigen presentation is the process by which certain cell in the body especially antigen ...
The Major Histocompatibility complex (MHC) system known as the human leukocyte antigen (HLA) in humans is located on the short arm of chromosome 6 (6p21.3) and contains the most polymorphic gene cluster of the entire human genome. Furthermore, the HLA consists of three regions which have been designated as class I, class II, and class III based on the structure and function of gene products ...
Abstract | Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high- affinity antibodies without T cell help. CD4 T cells, which provide. such ...
Antigen Presentation and Major Histocompatibility Complex. Pavel P. Nesmiyanov, in Encyclopedia of Infection and Immunity, 2022 Introduction. Antigen presentation is a process of displaying parts of antigenic fragments—epitopes—to the immune cells bearing corresponding antigen receptors. Antigen can be presented by any nucleated cell of the organism, but there is a specific subset of cells ...
pathway. This process requires the chaperone HLA-DM, and, in the case of B cells, the HLA-DOmolecule. MHC class II molecules loaded with foreign peptide are then transported to the cell membrane to present their cargo to CD4+ T cells. Thereafter, the process of antigen presentation by means of MHC class II molecules basically
Antigen presentation is the initial stage of the immune response and is mainly a process in which antigen-presenting cells (e.g., dendritic cells (DCs), macrophages) take up and process them into antigenic peptides so that they can be recognized by immunocompetent cells. Allergologia et Immunopathologia, 2019. About this page.
Two types of antigens are processed by cells for presentation on the cell surface. endogenous antigens are proteins produced by the cell. exogenous antigens are proteins that are taken up by the cell. Both types are linked to major histocompatability complexes (MHC) during processing so that. they can be stably exported to the cell surface.
The immune system recognizes the exogenous antigen, initiates the inflammatory response and the subsequent steps leading to the production of specific antibodies by the B cells. 2 In human cells, the antigen presentation process is performed by the MHC I and II, and this mechanism is essential for the cell‐mediated immunity. 3 The MHC I is a ...
Step 6: display of MHC molecules at the cell surface. Most of the steps of antigen processing and presentation out-lined so far are constitutive in cells that express MHC. molecules. This has ...
Antigen presentation is a process in the body's immune system by which macrophages, dendritic cells and other cell types capture antigens, then present them to naive T-cells. The basis of adaptive immunity lies in the capacity of immune cells to distinguish between the body's own cells and infectious pathogens. The host's cells express ...
Antigen processing and presentation is the mechanism by which whole antigens are degraded and loaded onto MHC molecules for display on the cell surface for recognition by T cells. Both macrophages ...
Abstract. Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high-affinity antibodies without T cell help. CD4 + T cells, which provide such help, use antigen-specific receptors that recognize major histocompatibility complex (MHC) molecules in complex with peptide cargo.
Antigen processing, or the cytosolic pathway, is an immunological process that prepares antigens for presentation to special cells of the immune system called T lymphocytes.It is considered to be a stage of antigen presentation pathways. This process involves two distinct pathways for processing of antigens from an organism's own (self) proteins or intracellular pathogens (e.g. viruses), or ...
Antigen presentation by B2 cells primarily occurs through the BCR, ... These authors conclude that DC can process and present antigen similarly, irrespective of the antigen entry mechanism, whereas in the case of non-DC APCs, the entry mechanism can have a profound effect on antigen presentation . While in this report the authors studied ...
Figure 2. Trafficking of antigens for processing and presentation with MHC molecules: basic pathways and exceptions to the "rules". Cytosolic proteins are processed primarily by the action of the proteasome. The short peptides are then transported into the ER by TAP for subsequent assembly with MHC-I molecules.
This process is often time-consuming and can lead to T cell exhaustion, limiting the therapeutic potential of adoptive cell therapy. 2. ... The described approach with APC-mimic offers several advantages as an antigen-presentation system. As an antigen-agnostic approach, it can be used to present a wide range of antigens and is thus easily ...
This latter process generates an antigen-specific signal that is elicited through the binding of the TCR to the antigen-MHC I complex — commonly referred to as signal 1 in the immunology ...
The presentation of antigens by B cells on MHC class II molecules is a complex process that involves several stages: first, external antigens are recognized and captured by B cells through their B ...
This process is referred to as thymic B cell licensing, ... although the determinants of intrathymic antigen presentation that we have discussed here have primarily been established in mice, it is ...
Antigen Presentation to T Lymphocytes. In an adaptive immune response, antigen is recognized by two distinct sets of highly variable receptor molecules—the immunoglobulins that serve as antigen receptors on B cells and the antigen-specific receptors of T cells. As we saw in Chapter 3, T cells recognize only antigens that are displayed on cell ...
On February 8, 2001, FDA approved its first HBV antigen assay (DiaSorin's ETI-EBK PLUS) for use in the qualitative detection of hepatitis Be antigen (HBeAg) in human serum or plasma (ethylenediaminetetraacetic acid (EDTA), citrate, or heparin) as indicative of a laboratory diagnosis of HBV infection through its PMA process under section 515 of ...