
Business Brief
Medical device manufacturers continue to work towards achieving compliance with two new guidelines: European Medical Device Regulation (EU) 2017/745 and In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746 , or MDR and IVDR respectively. The MDR went into effect on May 26, 2021, making it critical for device makers who are not yet in compliance to address the new regulatory requirements outlined in this highly detailed and more rigorous regulation. A number of points in the MDR are known to cause issues for manufacturers, including revised rules around literature reviews, clinical evaluation reports, product unique device identifier (UDI) data, and post-market surveillance.
For literature reviews, the MDR calls for manufacturers to demonstrate that they have performed a robust justification to launch a device and keep it on the market.
This is achieved by producing a thorough literature search and retrieving adequate clinical data. The requirements for literature reviews, which are used to support clinical evaluation reports (CERs), are outlined in Annex XIV, Part A of Regulation (EU) 2017/245 .
For post-market surveillance, the MDR requires manufacturers to demonstrate that they have a proactive and systematic process in place to collect and utilize product-related information once medical devices are being commercialized. It is important to note that this process is continuous, part of the normal lifecycle of maintaining a device on the market. Guidelines for reporting frequencies are based on device risk class specifications and can be found in the MDR, as laid out in Chapter VII, Articles 83-92 as well as Annex III, Section 1 of Regulation (EU) 2017/245 .

Achieving MDR-Compliant Literature Reviews for Medical Devices
The MDR regulation outlines specific requirements for the literature review — a robust process of executing evidence-based research queries that describe medical device product safety, benefit, and risk. Literature reviews are comprehensive searches of published information found in scientific studies related to medical devices on the market. Appropriate search criteria play a critical role in deriving refined and relevant search results for the device in question. Ultimately, the literature review supports CERs and post-market clinical follow-up (PMCF) reports. The CER and PMCF reports are outlined in MDR Annex XIV, Parts A and B , respectively.
Challenges exist for medical device manufacturers performing literature reviews. As mentioned earlier, successful literature reviews depend on the proper framing and subsequent refinement of search criteria. For manufacturers facing compliance requirements for the first time, the literature review introduces new and unfamiliar processes that can change how manufactures procure, collect, and manage information. In addition, new regulatory requirements for literature reviews introduce workload and resource constraints, both of which can impact device readiness project timelines. Literature reviews present a specific obstacle as manufacturers need to demonstrate that they have kept accurate, rigorous, reproducible, and transparent records while continuously monitoring for adverse events and reporting any issues. Finally, the manual literature review is a time-consuming process, with each step in the lifecycle capable of delaying project milestones. More importantly, manual processes can potentially introduce significant errors into regulatory submissions, which may result in costly market delays.
The Medical Device Coordination Group (MDCG) has indicated that MEDDEV 2.7/1 Rev 4 is the key reference document providing guidance and best practices for literature reviews and the literature search protocol. MDCG 2020-13, Section D serves as a complement, providing guidelines on how to conduct the literature review. When possible, manufacturers should consider automating aspects of their literature reviews in order to save time and improve compliance rates.
The use of automated software solutions such as DistillerSR can eliminate many of the manual processes while providing transparent and audit-ready reviews.
Requirements, Challenges, and Best Practices for Post-Market Surveillance (PMS)
Philips Achieves Faster, More Accurate Literature Reviews for CER Submissions with DistillerSR. Read the case study here.
For each medical device that is put on the market, a Post-Market Surveillance (PMS) system must be maintained as part of the quality management system (QMS). The post-market surveillance plan consists of:
- a PMS procedure
- a Post-Market Surveillance Report (PMSR) or Periodic Safety Update Report (PSUR), based on the device risk class
The PMS procedure is one or more procedures that medical device manufacturers must create to establish their PMS system. The structure of these framework documents is largely left up to manufacturers but will often take the form of work instructions or standard operating procedures (SOPs). When implementing PMS procedures, manufacturers will likely find a number of additional SOPs internally that will be affected and must be changed. As a result, device makers should consider the broader QMS repercussions of updating their PMS procedures when seeking MDR compliance.
Ultimately, the PMS procedure aims to provide feedback on the benefit-risk determination, clinical evaluation, usability, safety and clinical performance, reportable trends, technical documentation, and post-market surveillance, and when necessary, medical device manufacturers should consider corrective actions for the device in question. Much of this feedback can be sourced through scientific literature.
The PMS plan, outlined in Chapter VII, Article 84 , is a comprehensive document that defines the processes and methods involved in collecting and analyzing data related to product performance.
This plan establishes a rigorous, proactive, and systematic process to characterize device performance and compare it to similar medical devices on the market, as well as outlining how data is to be collected. The post-market surveillance plan applies to all device risk classes and should be updated when necessary, though updates can be performed internally and do not need to be made available to notified bodies except upon request for the information.
The PMS plan is part of the MDR technical documentation and outlines the PMS procedure(s) outlined above, as well as the PMSR and the PSUR.
Medical device manufacturers must include in the PMS plan market feedback and customer feedback and complaints, as well as product vigilance, recalls, and literature reviews (including database and register reviews). As a result, the PMS plan will impact multiple other QMS records, including the CER, risk management procedures, corrective and preventive action (CAPA) procedures, trend reporting, PMCF, and instructions for use (IFUs). The MDCG does not provide a solid guidance document or template upon which manufacturers can base their PMS plan. As a result, manufacturers must work to closely follow the MDR guidelines and establish their own planning procedure.
One area of specific confusion among medical device manufacturers unfamiliar with MDR is the revised requirements for PMCF, PMSR, and PSUR.
These documents are distinct from one another and may only apply to specific device risk classifications; however, they do sound similar and can be easily confused with one another.
The PMCF is outlined in Annex XIV, Part B . The PMCF is a specific and proactive form of post-market surveillance required for all Class IIb and Class III medical devices that summarizes clinical evidence from actual and similar devices on the market, including literature publications that highlight product safety and performance. The PMCF consists of both a PMCF plan and a PMCF report. The best practice, in compliance with PMCF requirements, is to follow the guideline documents published by the MDCG, MDCG 2020-07 (PMCF plan) and MDCG 2020-08 (PMCF evaluation report).
Another area of uncertainty for medical device manufacturers relates to the PMSR and the PSUR and their reporting frequency requirements.
The requirements are briefly outlined in Chapter VII, Article 85 . This item summarizes results and conclusions of the PMS data, describes corrective actions taken when applicable, and includes both reactive (i.e., complaint-based) as well as proactive (i.e., PMCF) post-market surveillance practices.
The PMSR is only required for Class I devices and becomes part of the technical documentation (TD).

A PMSR is only submitted to competent authorities upon request, rather than being made available for notified bodies during conformity assessment reviews or through EUDAMED. In addition, medical device manufacturers are responsible for updating the PMSR only when necessary.
The PSUR requirements are outlined in Chapter VII, Article 86 . This document is similar to the PMSR but applies only to Class IIa, IIb, III, and implantable devices. It also summarizes the results and conclusions of the PMS data and post-market information, vigilance reports, corrective actions, and status of devices on the market.
The PSUR, however, must be updated at least every two years for Class IIa devices and Class IIb non-implantables, and at least every year for Class IIb implantable and Class III devices. In addition, PSURs are submitted during notified body (NB) conformity assessment reviews for Class IIa and Class IIb non-implantable devices, and submitted via EUDAMED for notified body reviews for Class IIb implantables and Class III devices.
It is important to note that while the European Commission does not specify formats for each of these components, there are guidance documents provided by the MDCG that can assist manufacturers with how best to implement these MDR requirements.
Best Practices and Strategies For Compliant EU MDR Literature Reviews
Literature review and post-market surveillance items overlap at two key points during MDR remediation.
The first is in CERs, as outlined in Chapter VI and Annex XIV, Part A . CERs combine key findings in literature reviews and post-market surveillance to summarize clinical evidence and non-clinical data that support device performance and safety.
The second overlap is in PMCF, as outlined in Annex XIV, Part B . PMCFs serve as inputs into CERs and address the post-market surveillance plan by providing evidence of continuous collection of clinical evidence.
PMCFs include data from literature publications on device safety and performance as well as adverse events reports. The literature review can be broken down into a lifecycle, with each step employing unique strategies for ensuring compliance with regulatory requirements:

Define the Research Question
Defining an appropriately scoped research question is a critical component of the literature review. Research questions that are too broad may lead to a broad set of search criteria and return too many references that will need to be qualified. Overly narrow research questions may fail to retrieve all relevant evidence- based data and capture the full complexity and function of the medical device product. Furthermore, the research question must yield search results that support the requirements of clinical data as laid out in EU 2017/745 Annex XIV, Part A, Sections 1-4 . Specifically, clinical data from the literature review process that supports the CER must include both favorable and unfavorable data and must support the technical, biological, and clinical characteristics of the device.
Defining the research question is the first step in performing a literature summary, but it is also a step that is revisited throughout the literature review lifecycle. MEDDEV 2.7/1 Revision 4, Appendix 5 recommends establishing a literature review protocol that can be used as a key audit tool that will contain the research question. As such, the protocol will define key search terms, databases and sources of data, selection criteria, appraisal and analysis plans, and other components that support the clinical evaluation and performance evaluation reports. Establishing a literature review protocol using this reference as guidance, and including each of these components combined with a well-defined research question, will set medical device manufacturers up to successfully comply with EU MDR regulatory requirements.
Search Relevant Databases
For example, healthcare-focused databases like EMBASE or PubMed should be used, in addition to non-EU-based safety databases such as MAUDE (Manufacturer and User Facility Device Experience) . The aim here is to identify the most robust and directly relevant data that supports the safety and efficacy of the medical device product.
Screen References for Relevance
Retrieve full-text articles, extract and appraise the quality of the data.
Methods for appraising the quality of the data should be registered in the literature review protocol, as outlined in MEDDEV 2.7/1 Revision 4, Annex 5, Section 3 . Poor data quality can result in one or more of the following scenarios:
- Lack of information on elementary aspects (disclosure omissions)
- Numbers too small for statistical significance
- Improper statistical methods
- Lack of adequate controls
- Improper collection of mortality and serious adverse events data
- Misinterpretation by the authors
- Illegal activities
Standardizing on a comprehensive assessment tool, such as those incorporated in literature review platforms, will ensure all potential data quality shortcomings can clearly be identified and tracked for auditability. DistillerSR comes with a number of industry-standard assessment templates that can be adopted and modified to meet your specific literature review protocol while automatically tracking and associating all decisions and data collected to the appropriate article.
Document Purpose and Methods
The purpose and methods should all be documented in the literature review protocol, as outlined in MEDDEV 2.7/1 Revision 4, Annex 5, Section 3 . A robust, easily traceable literature review protocol will ensure repeatability and transparency in audits, increasing the likelihood of regulatory compliance. In addition, the protocol can serve as a project management guiding document to drive resources supporting the literature review process.
Literature review platforms help to facilitate adherence to specific protocols while automatically tracking each action. DistillerSR, for example, makes it easy to view the provenance of every cell of data and ensures complete transparency and auditability of the entire process, which enables a fully defensible, transparent, and repeatable review.
Monitor for New Literature and Adjust the Research Question, If Necessary
Learn How Literature Review Automation Improves CER and PER Program Management. Read the business brief here.
Medical Device Industry Trends and Opportunities
Manufacturers must follow the regulatory requirements laid out in the EU-MDR 2017/245, along with industry guidelines provided by MDCG, to make their medical device literature reviews and post-market surveillance programs MDR compliant. Manufacturers that have a larger ecosystem may wish to impose different strategies than smaller companies with fewer products.
One strategy employed by smaller companies simply looking to avoid audit findings and efficiently reach MDR regulatory compliance has been to integrate multiple product lines into the same CER. This must be done strategically to ensure that different devices still meet the MDR requirements for equivalence in technical, clinical, and biological device characteristics, and to ensure auditors can be provided with a rationale for how each product meets standards for the same generic device grouping. The MDR defines a generic device group in Article 2(7) as “a group of devices with the same or similar intended uses or with technological similarities that can be classified even without considering specific features.”
The guidance documents MDCG 2019-13, ISO 13485 , and EU Implementation Directive (EU) 2017/2195 provide additional clarity.
Larger companies can also employ these tactics to consolidate larger product families in CERs. Again, establishing technical, clinical, and biological equivalence for each device SKU is critical to justifying these groupings.
Standardization Is Critical to Achieving Compliance with Your Post Market Surveillance Plan
When putting PMCF and PMS requirements into practice, all manufacturers can leverage templates and guidelines that are published by the European Commission and MDCG. For example, ensuring that a compliant PMS plan is in place can be achieved by writing policies that heavily mirror the EU MDR 2017/245. The use of templates can provide a repeatable, consistent, and transparent process to standardize PMCF, PMSR, and PSUR reports across the business. Each can be customized for different product families as needed. Moreover, standardizing literature review processes on a software platform such as DistillerSR further establishes consistency through automated processes, drives internal efficiencies, and ensures all activities are traceable and auditable.
Meeting the regulatory requirements for both literature reviews and post-market surveillance can be challenging for many companies. However, by doing so, manufacturers can ensure that they are well on their way to fully achieving MDR compliance.
Ultimately, leveraging the right tools to do some of the heavy lifting throughout the literature review process and optimizing workflows for continuous data monitoring will be critical for continuous compliance with the EU MDR and, in May 2022, the EU IVDR. Learn more about how do you conduct post-marketing surveillance .
Download the PDF version of this solution brief.
Related resources.

Philips achieves faster, more accurate literature reviews for CER submissions with DistillerSR.
Read Case Study

Learn More About DistillerSR
Literature Reviews for Medical Devices: 6 Expert Tips
Table of Contents
Catarina Carrão , freelance medical writer on Kolabtree, outlines the importance of literature reviews for medical devices and best practices to follow.
A clinical evaluation is an ongoing process, conducted throughout the life cycle of a medical device . It is usually first performed during the development phase of the medical device, in order to identify the data that it needs to be granted market access. In the European Union, for an initial CE-marking, a Clinical Evaluation Report (CER) is mandatory, and it must be actively updated continuously afterwards[ 1] . In the United States, a Pre-Market Approval (PMA ) [2] is the Food and Drug Administration (FDA) process for scientific and regulatory review, to evaluate the safety and effectiveness of a medical device (Class III), so that it can reach the consumer. It also uses an evidence-based review system for scientific evaluation of medical devices.
This process of clinical evaluation is fundamental, because it ensures the safety and performance of the device based on abundant clinical evidence, throughout the lifetime of the medical device on the market. It enables Notified Bodies (NBs) and Competent Authorities to read through the clinical evidence to demonstrate the conformity of the device with the essential requirements, not just for initial marketing, but throughout its lifetime ( e.g. , fulfilment of post- market surveillance and reporting requirements) [ 1] .
Literature Reviews for Medical Devices
Literature reviews are crucial to the success of a CER and PMA, because a solid and systematic literature research strategy fortifies every stage of the medical device life cycle process: from concept and design, through clinical trials to release of the medical device and reimbursement [ 3] . So, more than just a wise investment, the screening of the literature to comply with regulatory authorities during the approval process and for post-market surveillance, is fundamental to the global success of any marketed medical device.
For many companies, especially Small and Medium Enterprises (SMEs), the data retrieved from literature searches will represent most, if not all, of the data collected. As such, this search identifies sources of clinical data for establishing the current knowledge or “the-state-of-the-art” that describes the clinical background in the corresponding medical field; the clinical data that is relevant to the device under evaluation, or to an equivalent device (if equivalence is claimed in a CER 1 , or 510K[ 4] ); and, the identification of potential clinical hazards. That’s why it is so critically important to develop a literature search strategy that is robust, and can be replicated during subsequent updates by any person.
1. Search protocol (Stage 1)
The searching strategy should be thorough and objective, i.e. it should identify all relevant favourable and unfavourable data; and, should be carried out based on a search protocol [ 1] . The search protocol documents the planning of the search before execution. Once the searches have been executed, the adequacy of the searches should be verified, and a literature search report should be compiled to present details, with any deviations from the literature search protocol documented, together with the results of the search. It is important that the literature search is documented to such degree that the methods can be appraised critically, the results can be verified, and the search reproduced if necessary.
According to the regulations [ 1], [2] , the literature search protocol should include the following elements [ 5] :
- Sources of data used ( e.g. , MEDLINE/PubMed, Embase, Google Scholar, ResearchGate, internet searches, etc.);
- The methodology used for the searches;
- The exact search terms and parameters used to search scientific databases ( e.g. , dates);
- Specific selection or exclusion criteria along with justifications for each;
- How was duplication of data from multiple sources addressed;
- How was data integrity ensured ( e.g. , Quality Control Methods or second reviewers);
- How each data source was appraised, and its relevance for the specific device;
- Analysis and data processing handling.
The search strategy must be broad enough to ensure that no essential information is missed, but still allow precise identification of relevant results. This can involve the use of search features such as filters to narrow down the result set; sub-headings based on key concepts such as device adverse effects, or device comparison; and triage and analysis methods to identify the most relevant literature. The results themselves are generally in the form of a list of citations or data, with descriptive indexing tags and other key information.
Abstracts lack sufficient detail to allow issues to be evaluated thoroughly and independently, but may be sufficient to allow a first evaluation of the relevance of a paper [ 1] . Good research informatics solutions allow both flagging of the citation and annotation of the article text, so that teams can work closely on individual items [ 3] . Copies of the full text papers should be included in the final files. The literature search protocol(s), the literature search report(s), and full text copies of articles and relevant documents, become part of the final technical documentation for the medical device.
2. Possible errors
A precise literature search provides accurate evidence; but, unless implemented correctly, the result can be misleading, time consuming, or even useless [ 6] . There are errors related to the volume of evidence, relevance of the data, tone of evidence, and its value to the research topic, that might undermine even a high-skilled researcher. It is necessary to focus the literature search on precise topics, and obtain relevant evidence within a stipulated time, otherwise outcomes might deviate.
Usually, errors have their origin in an incorrect use of primary attributes of literature search, viz. , keywords, Boolean, and database [ 6] . For example, the evaluator could create errors in setting eligibility criteria (type of literature and databases); or errors in selecting keywords and Boolean logics; or even, errors in setting up search phrases in the database.
These attributes can lead to errors of inclusion (too much data, partly not relevant to the issue); or, exclusion of important data, because of too stringent keyword use. But it can also lead to errors of “inclusive exclusions”, due to bias by literature professionals in the searches; and, “exclusive inclusions”, with the use of highly specific key-terms with inadequate Booleans, or even the exclusion of synonyms for the same medical terminology. An error of “exclusive exclusions“ is also called the „error of limited relevance“, and it happens in many cases. This error is a combination of bias and specific exclusiveness; where the search phrases constructed will be biased to only one-sided data trends, and the terms selected will be too exclusive to return sufficient information [ 6] .
Besides possible errors in retrieving important clinical data, uncertainty of the final literature review also arises from two sources: the methodological quality of the data, and the relevance of the data to the evaluation of the device in relation to the different aspects of its intended purpose [ 1] . Both sources of uncertainty should be analysed, in order to determine a weighting for each data set. As such, a balanced assessment of the quality of the data is essential to the success of the literature review search.
Also read: Clinical evaluation for EU MDR Compliance: 5 Dos and Don’ts
3. Appraisal of the clinical data
When appraising the data generated by the database search (Stage 2), the evaluator is looking to make sure it has statistically significant data sets, uses proper statistical methods, has adequate controls, and properly collects mortality and/or serious adverse event data. It is essential that the correct assessment is done based on the complete text of the publications found, not just by reading the abstracts or summaries. For each document appraised, there needs to be a documentation of the appraisal to the point that it could be reasonably reviewed by others. The appraisal results should also support conclusions about the clinical safety and performance of the finished device ( e.g. , citing non-device-related literature would be ranked low for appraisal) 5 . There are some red flags provided by the regulation in order to appraise the medical publications, for example:
- The article lacks basic information such as the methods used, number of patients, identity of products, etc.;
- Has data sets that are too small to be statistically significant;
- Contains data that applies improper statistical methods;
- Employs studies that lack adequate controls;
- Has an improper collection of mortality and serious adverse event data;
- Depicts a misrepresentation by the authors;
The evaluators should verify whether clinical investigations have been defined in such a way as to confirm or refute the manufacturer’s claims for the device; and, whether these investigations include an adequate number of observations to guarantee the scientific validity of the conclusions [ 1] . Some papers considered unsuitable for demonstration of adequate performance because of poor elements of the study design or inadequate analysis, may still contain data suitable for safety analysis, or vice versa.
Typically, clinical data should receive the highest weighting, when generated through a well designed and monitored randomized controlled clinical investigation (also called randomised controlled trial), conducted with the device under evaluation in its intended purpose, with patients and users that are representative of the target population [ 1] . It is acknowledged by the regulators that randomized clinical investigations may not always be feasible and/or appropriate, and the use of alternative study designs may provide relevant clinical information of adequate weighting. When rejecting evidence, the evaluators should document the reasons.
4. Analysis and conclusions generated from the clinical data
During the analysis stage (Stage 3), a comprehensive assessment is done to determine if the data found actually meets the clinical safety requirements, clinical performance requirements, and General Safety and Performance Requirements (GSPR). It is important to evaluate if the risk-benefit ratio of the medical device is appropriate based on the intended purpose of the device, or if the device can actually achieve all performance claims made by the manufacturer. Also, if the materials supplied by the manufacturer (labelling/instructions) are adequate to describe the intended purpose and mitigate the risk [ 7] . All in all, the evaluation is intended to conclude whether the risks of the device are minimal and acceptable according to its purpose. As such, understanding the interaction between the device and body, the number and severity of adverse events, and the current standards of care, are some of the gaps that will need to be taken into account [ 1] .
The data from the literature is often put into Excel tables, which is a convenient way to compare different study details, patient populations, endpoints, adverse events, etc. [ 7] . This is extremely helpful in noting differences between studies when writing the summary and conclusions. The evaluators should also include aspects such as rare complications, uncertainties regarding medium- and long-term performance, or safety under wide-spread use; and, identify additional clinical investigations, or other measures, that are necessary in order to generate any missing data [ 1] .
5. Informatic tools
This massive task of literature search can be streamlined with the right research informatics solutions. Nowadays, the key is to select a literature database with appropriate medical device coverage, in terms of content and indexing. In a case study reviewing literature about a particular medical device, the top research informatics solutions were Embase and Science Citation Index (SCI) [ 3]. But also, Medline, and BioMed Central are considered top research informatic solutions for retrieving medical device literature and can be used by anyone.
Using a solution that performs automated searches and notifies the user of relevant new data via email alerts or RSS feeds saves considerable time; and, keeps the evaluator updated until the final stage of submission [ 3] . If the tool has the appropriate indexing and tagging, this will simplify literature triage; and, more importantly, it dramatically reduces the risk of missing adverse event reports. The combination of a good literature search tool and a trained evaluator can be the best solution to avoid errors and limitations of literature review search.
Also read: Writing a Clinical Evaluation Report: 5 Quick Tips
6. Process flow
Modern regulatory requirements have made biomedical literature research an essential part of the medical device life cycle; as such, a good strategy to find and summarize all the relevant clinical data is a must:
- Identify the question to be answered;
- Decide which database better fits the question,
- Identify the Medical Subject Headings (MeSH), or Embase Subject Headings (EmTree);
- Recognize the correct Boolean terms to use;
- Create and document the Literature Search Protocol;
- Run the Literature search automation tool;
- Appraise and analyse the literature (tabulation),
- Summarize conclusions.
Conclusions
The future looks promising for the dauting task of doing a systematic literature review , since artificial intelligence and natural language processing based-tools with cognitive capabilities provide a near-perfect solution [ 6] . But, until then, an organized and highly-skilled research-evaluator is essential to execute a dedicated strategy for literature monitoring, triage and analysis.
Need help with literature reviews for medical devices? Hire clinical evaluation experts and literature search specialists on Kolabtree.
References:
- European Commission. CLINICAL EVALUATION: A GUIDE FOR MANUFACTURERS AND NOTIFIED BODIES UNDER DIRECTIVES 93/42/EEC and 90/385/EEC. MEDDEV 27/1 revision 4 . 2016.
- FDA. Premarket Approval (PMA). 2019;18th May 2020.
- Elsevier. BOOSTING THE SUCCESS OF MEDICAL DEVICE DEVELOPMENT WITH SYSTEMATIC LITERATURE REVIEWS. 2014;18th May 2020.
- FDA. Premarket Notification 510(k). 2020;19th May 2020.
- OrielStat. Creating an EU CER Literature Review Protocol and Reviewing Medical Device Clinical Data. 2019;18th May 2020.
- Ashish Indani* SRB, Nadeem Ansari. Literature Search for Scientific Processes in Medical Devices: Challenges, Errors, and Mitigation Strategies. Tata Consultancies . 2017;7.
- OrielStat. Analyzing Your Medical Device Clinical Datasets and Drawing Conclusions. 2019;18th May 2020.
Unlock Corporate Benefits • Secure Payment Assistance • Onboarding Support • Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.
About Author
Ramya Sriram manages digital content and communications at Kolabtree (kolabtree.com), the world's largest freelancing platform for scientists. She has over a decade of experience in publishing, advertising and digital content creation.
Related Posts
How on-demand fda submission consultants can prove valuable to your business, three ways on-demand medical writers can help your business , fda 510k submissions guide: free kolabtree whitepaper, leave a reply cancel reply.
Save my name, email, and website in this browser for the next time I comment.
Automated page speed optimizations for fast site performance
Advertisement
Medical device usability: literature review, current status, and challenges
- Published: 15 February 2020
- Volume 36 , pages 163–170, ( 2020 )
Cite this article
- Marylene Sousa Guimarães Roma 1 &
- Euler de Vilhena Garcia ORCID: orcid.org/0000-0002-6357-4701 2
938 Accesses
4 Citations
Explore all metrics
User involvement during medical device (MD) development and usability engineering techniques may help reduce serious adverse events due to human error during MD use. This paper reviews the scientific literature on MD usability and critically analyzes the MD design and development (MDDD) process.
Literature review
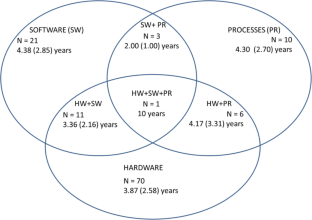
We searched 20 international databases for papers on usability and MDs. After applying exclusion criteria and removing duplicates, we analyzed 144 scientific papers regarding usability aspects and evaluated the target audience and study scope. Among hardware (HW), software (SW), and process (PR) evaluation methods, HW was the most evaluated (49% of papers), while the remainder analyzed HW + SW (15.2%), HW + PR (4.2%), and HW + SW + PR (0.7%). Task analysis, scenario simulation, and questionnaires were the most commonly used techniques (31.6%, 18.4%, and 12.8%, respectively). The target audiences were primarily patients/lay users (62%) and medical staff (14%). Gastroenterology (16.7%), nuclear medicine (13%), and nephrology/urology (9.3%) were the most referred specialties. We found that 48% of all papers did not mention any health facility or service analyzed, while 25.3% analyzed homecare services. Considering the usability scope, product evaluation (32%) and verification or validation trials (29%) were the most common.
Usability in MDDD
We present a brief review of the MDDD scenario and argue that better selection of usability methodologies in MDDD should be based around three factors: application of current technical standards on usability, usage of health technology assessment literature, consideration of ethics-related specificities of MD design.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

Clinical Usability Studies – Clash of Cultures? Study Design Proposal from Lessons Learned

Usability Testing Medical Devices: A Practical Guide to Minimizing Risk and Maximizing Success

MeD UD – A Process Reference Model for Usability Design in Medical Devices
ABNT. Associação Brasileira de Normas Técnicas. ISO/IEC 9126–1: engenharia de software: qualidade de produto. Parte 1: modelo de qualidade. Rio de Janeiro: ABNT; 2003.
Google Scholar
ABNT. Associação Brasileira de Normas Técnicas. ABNT NBR ISO 14971:2009: Produtos para a saúde — Aplicação de gerenciamento de risco a produtos para a saúde. Rio de Janeiro: ABNT; 2009.
ABNT. Associação Brasileira de Normas Técnicas. NBR ISO 9241-11: requisitos ergonômicos para o trabalho com dispositivos de interação visual. Parte 11: orientações sobre usabilidade. Rio de Janeiro: ABNT; 2011a.
ABNT. Associação Brasileira de Normas Técnicas. NBR ISO 9241-210: ergonomia da interação humano-sistema. Parte 210: projeto centrado no ser humano para sistemas interativos. Rio de Janeiro: ABNT; 2011b.
ABNT. Associação Brasileira de Normas Técnicas. ISO/TR 16982: ergonomia da interação humano-sistema: métodos de usabilidade que apoiam o projeto centrado no usuário. Rio de Janeiro: ABNT; 2014.
ABNT. Associação Brasileira de Normas Técnicas. ABNT NBR IEC 62366:2010 Emenda 1:2016 Produtos para a saúde - Aplicação da engenharia de usabilidade a produtos para a sáude. Rio de Janeiro: ABNT; 2016.
ANVISA. Manual de tecnovigilancia: abordagens de vigilância sanitária de produtos para a saúde comercializados no Brasil. Brasília: Ministério da Saúde; 2010.
ANVISA. Diretrizes metodológicas: elaboração de estudos para avaliação de equipamentos médicos assistenciais. Brasília: Ministério da Saúde; 2013a.
ANVISA. Relatório do Quantitativo de notificações/Número de notificações por mês de eventos adversos. Brasília: Ministério da Saúde; 2013b.
ANVISA. Resolução da Diretoria Colegiada – RDC n° 16, de 28 de março de 2013. Aprova o Regulamento Técnico de Boas Práticas de Fabricação de Produtos Médicos e Produtos para Diagnóstico de Uso In Vitro e dá outras providências. Brasília: Ministério da Saúde; 2013c.
ANVISA. Resolução da Diretoria Colegiada – RDC n° 15, de 20 de fevereiro de 2015. Dispõe sobre o regulamento para a realização de ensaios clínicos com dispositivos médicos no Brasil. Brasília: Ministério da Saúde; 2015.
Auer A, Jarmai K. Implementing responsible research and innovation practices in SMEs: insights into drivers and barriers from the Austrian medical device sector. Sustainability. 2017;10(1):17–35.
Article Google Scholar
Bellido D, Leon A, Manas M, Marchan E, Esquinas G, Ros J. Adverse events in an internal medicine: a prospective study. Rev Calid Asist. 2017. https://doi.org/10.1016/j.cali.2017.02.003 .
BRASIL. Diretrizes metodológicas: elaboração de pareceres técnico-científicos. Brasília: Ministério da Saúde; 2014a.
BRASIL. Documento de referência para o Programa Nacional de Segurança do Paciente. Brasília: Fundação Oswaldo Cruz; 2014b.
Brown A, Dixon D, Eatock J, Meenan B, Young T. A survey of success factors in new product development in the medical devices industry. IEMCE. 2008. https://doi.org/10.1109/IEMCE.2008.4617987 .
De Falco I, Tortora G, Dario P, Menciassi A. An integrated system for wireless capsule endoscopy in a liquid-distended stomach. IEEE Trans Biomed Eng. 2014. https://doi.org/10.1109/tbme.2013.2290018 .
Doorn N, Fahlquist J. Responsibility in engineering: toward a new role for engineering ethicists. Bull Sci Technol Soc. 2010;30(3):222–30.
Dreyer M, Chefneux L, Goldberg A, von Heimburg J, Patrignani N, Schofield M, et al. Responsible innovation: a complementary view from industry with proposals for bridging different perspectives. Sustainability. 2017. https://doi.org/10.3390/su9101719 .
Eindhoven D, Borleffs C, Dietz M, Schalij M, Brouwers C, de Bruijne M. Design and reliability of a specific instrument to evaluate patient safety for patients with acute myocardial infarction treated in a predefined care track: a retrospective patient record review study in a single tertiary hospital in the Netherlands. BMJ Open. 2017. https://doi.org/10.1136/bmjopen-2016-014360 .
EngelbergCenter. Biomedical innovation: identifying challenges and prioritizing needs for medical device innovation. In: Engelberg Center for Health Care Reform. 2014. https://www.brookings.edu/events/biomedical-innovation-identifying-challenges-and-prioritizing-needs-in-medical-device-research-and-development/ . Acessed 15 Dec 2018.
FDA. FDA Adverse Event Reporting System (FAERS) Statistics. 2014. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm070093.htm . Accessed 10 Nov 2014.
FDA. Framework for FDA’s Real-World Evidence Program. 2018. https://www.fda.gov/media/download&usg=AOvVaw0EVMNjhhH4ZQjqw1bLX3dv . Acessed 12 Apr 2019.
Flewwelling C, Easty A, Vicente K, Cafazzo J. The use of fault reporting of medical equipment to identify latent design flaws. J Biomed Inform. 2014. https://doi.org/10.1016/j.jbi.2014.04.009 .
Freund Y, Goulet H, Leblanc J, Bokobza J, Ray P, Maignan M, et al. Effect of systematic physician cross-checking on reducing adverse events in the emergency department: the CHARMED cluster randomized trial. JAMA Intern Med. 2018;178(6):812–9.
Fries R. Reliable design of medical devices. New York: CRC Press; 2006.
Fries J, Spitz P, Kraines R, Holman H. Measurement of patient outcome in arthritis. Arthritis Rheum. 2005;23:137–45.
Fujita S, Iida S, Nagai Y, Shimamori Y, Koyano K, Moriyama Y, et al. Estimation of the number of patient deaths recognized by a medical practitioner as caused by adverse events in hospitals in Japan: a cross-sectional study. Medicine (Baltimore). 2017. https://doi.org/10.1097/md.0000000000008128 .
Gurses A, Onzpk A, Pronovost P. Time to accelerate integration of human factors and ergonomics in patient safety. BMJ Qual Saf. 2011;21:347–51.
Hani S, Marcellis-Warin N. Open innovation and involvement of end-users in the medical device technologies design & development process: end-users perspectives. Technol Invest. 2016;7(3):73–85.
Hofmann B. Medicalization and overdiagnosis: different but alike. Med Health Care Philos. 2016;19(2):253–64.
Ijzerman MJ, Steuten LM. Early assessment of medical technologies to inform product development and market access: a review of methods and applications. Appl Health Econ Health Policy. 2011;9(5):331–347. https://doi.org/10.2165/11593380-000000000-00000 .
Jolly J, Hildebrand E, Branaghan R. Better instructions for use to improve reusable medical equipment (RME) sterility. Hum Factors. 2013;55(2):397–410.
Kangas M, Konttila A, Lindgren P, Winblad I, Jamsa T. Comparison of low-complexity fall detection algorithms for body attached accelerometers. Gait Posture. 2008;28(2):285–91.
L’Astorina A, Fiore MD. A new bet for scientists? Implementing the Responsible Research and Innovation (RRI) approach in the practices of research institutions. Relations. 2017. https://doi.org/10.7358/rela-2017-002-last .
Landman AB, Redden L, Neri P, Poole S, Horsky J, Raja AS, et al. Using a medical simulation center as an electronic health record usability laboratory. J Am Med Inform Assoc. 2014;21(3):558–63.
Lehoux P, Miller F, Hivon M, Demers-Payette O, Urbach D. Clinicians as health technology designers: two contrasting tales about user involvement in innovation development. Health Policy Technol. 2013;2:122–30.
Leite C, Reis C, Binsfeld P, Rosa S. Novas Tecnologias Aplicada à Saúde: Desenvolvimento de Sistemas Dinâmicos – Conceitos, Aplicações e Utilização de Técnicas Inteligentes. Mossoró-RN:EDUERN; 2019.
Lynch R, Farrington C. Quantified lives and vital data. Exploring health and technology through personal medical devices. London: Palgrave Macmillan; 2018.
Book Google Scholar
Markiewicz K, Til J, IJzerman M. Medical devices early assessment methods: systematic literature review. Int J Technol Assess Health Care. 2014;30(2):137–46.
Markiewicz K, Til J, IJzerman M. Early assessment of medical devices in development for company decision making: an exploration of best practices. J Commer Biotechnol. 2017. https://doi.org/10.5912/jcb780 .
Martin JL, Clark DJ, Morgan SP, Crowe JA, Murphy E. A user-centred approach to requirements elicitation in medical device development: a case study from an industry perspective. Appl Ergon. 2012;43(1):184–90.
Matsumoto K, Nagahara A, Ueyama H, Konuma H, Morimoto T, Sasaki H, et al. Development and clinical usability of a new traction device “medical ring” for endoscopic submucosal dissection of early gastric cancer. Surg Endosc. 2013;27:3444–51. https://doi.org/10.1007/s00464-013-2887-6 .
McCrory B, Lowndes BR, Lagrange CA, Miller EE, Hallbeck MS. Comparative usability testing of conventional and single incision laparoscopic surgery devices. Hum Factors. 2013;55(3):619–31.
Mendes W, Pavao ALB, Martins M, Travassos C. The application of Iberoamerican study of adverse events (IBEAS) methodology in Brazilian hospitals. Int J Qual Health Care. 2018;30(6):480–5.
Money A, Barnett J, Kuljis J, Craven M, Martin J, Young T. The role of the user within the medical device design and development process: medical device manufacturers’ perspectives. BMC Med Inform Decis Mak. 2011. https://doi.org/10.1186/1472-6947-11-15 .
Santos ICT, Gazelle GS, Rocha LA, Tavares JMRS. Modeling of the medical device development process. Expert Rev Med Devices. 2012;9(5):537–43.
Sarah S, Jennifer M, Alexandra L, Michael C, Sonja ON, Julie B. Medical device design in context: a model of user–device interaction and consequences. Displays. 2012. https://doi.org/10.1016/j.displa.2011.12.001 .
Shah SGS, Robinson I. Benefits of and barriers to involving users in medical device technology development and evaluation. Int J Technol Assess Health Care. 2007;23(1):131–7.
Tarricone R, Torbica A, Drummond M. Challenges in the assessment of medical devices: the MedtecHTA project. Health Econ. 2017a. https://doi.org/10.1002/hec.3469 .
Tarricone R, Torbica A, Drummond M. Key recommendations from the MedtecHTA project. Health Econ. 2017b. https://doi.org/10.1002/hec.3468 .
Turchetti G, Spadoni E, Geisler E. Health technology assessment. IEEE Eng Med Biol Mag. 2010;29(3):70–6.
Van de Poel I, Asveld L, Flipse S, Klaassen P, Scholten V, Yaghmaei E. Company strategies for Responsible Research and Innovation (RRI): a conceptual model. Sustainability. 2017;9(11):2045.
Vincent C, Li Y, Blandford A. Integration of human factors and ergonomics during medical device design and development: It’s all about communication. Appl Ergon. 2014;45:413–9.
Walton M, Harrison R, Kelly P, Smith-Merry J, Manias E, Jorm C, et al. Patients reports of adverse events: a data linkage study of Australian adults aged 45 years and over. BMJ Qual Saf. 2017;26(9):743–50.
WHO. Health technology assessment of medical devices, Geneva. 2012.
Wiklund M, Kendler J, Strochlic A. Usability testing of medical devices. 2a ed. Boca Ration: CRC Press; 2011.
Download references
Acknowledgments
We would like to thank Editage ( www.editage.com ) for English language editing.
Author information
Authors and affiliations.
Biomedical Equipment Associate Course Coordination, Federal Institute of Brasilia at Ceilândia, QNN 26, Área Especial, Ceilândia, DF, Brazil
Marylene Sousa Guimarães Roma
Biomedical Engineering Graduate Program, University of Brasilia at Gama, Área Especial de Indústria Projeção A – Setor Leste, Gama, DF, Brazil
Euler de Vilhena Garcia
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Euler de Vilhena Garcia .
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Reprints and permissions
About this article
Roma, M.S.G., de Vilhena Garcia, E. Medical device usability: literature review, current status, and challenges. Res. Biomed. Eng. 36 , 163–170 (2020). https://doi.org/10.1007/s42600-019-00037-8
Download citation
Received : 05 April 2019
Accepted : 26 December 2019
Published : 15 February 2020
Issue Date : June 2020
DOI : https://doi.org/10.1007/s42600-019-00037-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Medical devices
- User-centered design
- Health technology assessment
- Find a journal
- Publish with us
- Track your research

More than a Quality Management System: Tools for the entire MedTech Lifecycle.
Featured Capabilities:

Experience the #1 QMS software for medical device companies first-hand. Click through an interactive demo.
Accelerate development with integrated design control and risk software.

Schedule a custom demo of Greenlight Guru Product now.
Data collection and management designed for MedTech clinical trials.

Get a personalized demo of Greenlight Guru Clinical today.
- By Initiative
- Migrating From Paper
- Managing and Assessing Risk
- Preparing for Regulatory Submissions
- Becoming Audit Ready
- Managing Postmarket Quality
- Small Business
- By Function
- Product / R&D
- ROI Calculator
- Customer Success
- Case Studies
- Checklists & Templates
- eBooks & Guides
- Content Hub
- Live & Virtual Events
- True Quality Roadshow
- Quality Pricing
- Clinical Pricing

The Clinical Evaluation Literature Review Process: Identifying and Appraising Clinical Data (Part 2 of 4)
.png "medical device literature review")
A successful clinical evaluation hinges on your ability to find and appraise the data you’ll need to demonstrate your device’s safety and performance.
However, it’s also one of the more difficult and time-consuming aspects of the clinical evaluation process. That’s why we’re devoting this article—part two of our four-part series on the clinical evaluation of medical devices—entirely to explaining the process behind gathering and appraising clinical data.
In this article, I’ll be covering the different types of clinical data, how to create a literature review protocol, and how to appraise the clinical data you find. We'll also cover how to prepare your Clinical Evaluation Report (CER) so it complies with MDR.
FREE DOWNLOAD: Click here to download your free PDF copy of our Clinical Evaluation Procedure Template.
Identify where you’ll obtain clinical data for your evaluation.
Your clinical evaluation will be based on both direct and indirect data.
Direct data is data that has been generated by your device, such as the data obtained from pilot studies or clinical trials. If your device is Class III or a Class IIb implantable, then you will be required to conduct clinical trials on your device and will generate direct data that way.
Direct data also includes registries and publicly available data on your device. You’ll also generate direct data from your postmarket surveillance activities , though this won’t be available to you during the initial clinical evaluation process.
Indirect clinical data is data that has been generated by an equivalent device, which is why it’s important to establish equivalence early on. The decision to claim equivalence (or not) will be one of the factors that affects your literature review.
The literature review is the means by which you obtain indirect clinical data. Many companies, especially if they are evaluating a low-risk device, will rely heavily on data from literature searches, as they won’t be carrying out any clinical trials.
The goals of your literature review
Your literature review will have two outputs:
Literature on your device and the equivalent device (if applicable).
A review of the “current knowledge/state of the art” necessary for appraising and analyzing the clinical data from the literature on your device and any equivalent devices.
Let’s talk about the term “state of the art” for a moment. MEDDEV 2.7.1 Rev 4 defines “state of the art” as the:
applicable standards and guidance documents, information relating to the medical condition managed with the device and its natural course, benchmark devices, other devices and medical alternatives available to the target population.
Your device does not live in a vacuum. The benefit-risk assessment of your device in part depends on what alternatives to your device exist, and whether they pose more or less risk to a patient than your device. Put simply, “state of the art” refers to the best practices currently available in the marketplace, and your clinical evaluation must take into account how your device compares to those alternatives.
Create your clinical evaluation literature review protocol
Once you know the outputs you’re looking for, it’s time to create a plan to obtain them. This means crafting a detailed protocol for your literature review. This isn’t an instance in which a Google search will turn up what you need; the literature review must be a systematic process.
Annex 5.3 of MEDDEV 2.7.1 Rev 4 includes a full list of what should be included in your search protocol, but here are a few pointers to keep in mind as you create your literature review protocol:
Your protocol will need to clearly define the objectives of your literature evaluation. For instance, if there are particular risks related to your device that have been identified, one of your objectives may be to search for literature containing information on that risk. On the other hand, you might also search for literature pertaining to any clear benefits that have been identified with your device.
You’ll need to designate specific terms that you’ll use in your search. If your terms are too broad, your search may return thousands of articles, many of which will be irrelevant to your clinical evaluation.
Your protocol also needs to establish which sources of information will be used for searches, and you need to be consistent about using those sources. That means applying the same search terms to several databases, such as PubMed , MEDLINE , or Embase .
Set up your search so that you get both titles and abstracts for the articles. The abstract will help you better determine whether you should acquire and read the full text of the article. This is what tends to make the literature review process so time-consuming—you need to read all of the relevant literature that your searches turn up.
Keep in mind : you cannot pick literature that will support your device and discard those papers that may be harmful to your cause. This is part of the reason you’re required to create a search protocol in the first place—it allows your Notified Body to retrace your steps if they have concerns about your literature review.
Appraise the clinical data from your literature review
Even a literature search with specific, defined terms can turn up hundreds of results. That’s why it’s important to have a systematic approach to appraising which results you should use and which you should discard. Generally, you want to consider four factors:
Suitability - Is this based on our device or an equivalent device?
Applicability - Is this about other devices that use the same technology?
Population - Is the population similar to the population we believe our device will treat?
Quality - Is this literature published in a peer reviewed journal? Is the data generated from a randomized double-blind trial with a placebo, for instance?
That last point, quality, is a big one. In MEDDEV 2.7.1 Rev. 4, Annex 6 offers a number of points for concern you should look out for including:
A lack of information on elementary aspects, such as methods, patient population, side-effects, or clinical outcomes.
Statistically insignificant data or improper statistical methods.
A lack of adequate controls leading to bias or confounding.
The improper collection of mortality and serious adverse events data.
Misinterpretation of data by the authors, such as when the conclusions they draw are not in line with the results section of the report.
Any illegal activities, such as clinical investigations that were not conducted in compliance with local regulations.
Once you’ve eliminated any results with these types of flaws, you’ll need to weigh the importance of the different data that you’ve obtained from your scientifically valid sources.
At its most basic level, this means assigning a higher weighting to high-quality data that is most relevant to your device, and assigning a lower weighting to less relevant data. If you’re unsure about how to weight your data sets, Appendix III of MDCG 2020-6 provides a suggested hierarchy for clinical data that should be helpful.
It’s important to note that the appraisal of data may require someone with a specialized skill set, such as a statistician. In fact, it’s highly unlikely that one QA/RA professional can handle an entire clinical evaluation by themselves, and I certainly wouldn’t recommend trying.
Finally, your appraisal of the data must be documented, and it should be presented clearly enough for a third party to review your decisions.
Up next: Stage 3 of clinical evaluation — analyzing your clinical data
Once you’ve gathered and appraised the data you need, the next step is analyzing it to determine whether or not it demonstrates compliance with clinical performance and safety requirements. Check out part three of our series for an in-depth look at the data analysis stage of your clinical evaluation .
Looking for an all-in-one QMS solution to advance the success of your in-market devices and integrates your post-market activities with product development efforts? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
Niki Price is a Medical Device Guru who has spent her entire career working with different types of medical devices. She began her journey in production, which is where she discovered how important and fulfilling this line of work was to her! Spending time in both Quality and R&D, she enjoys the product development...
Related Posts
Clinical evaluation of a medical device: creating a process and establishing equivalency (part 1 of 4), performing data analysis for your medical device’s clinical evaluation (part 3 of 4), what’s the difference between clinical evaluation and clinical investigation, subscribe to our blog.
Join 200,000+ other medical device professionals outperforming their peers.

Get your free eBook
The ultimate guide to clinical evaluation of a medical device in the eu.
%20The%20Ultimate%20Guide%20to%20Clinical%20Evaluation%20of%20a%20Medical%20Device-1.png?width=250&name=(cover)%20The%20Ultimate%20Guide%20to%20Clinical%20Evaluation%20of%20a%20Medical%20Device-1.png "medical device literature review")
- Checklists/Templates
- Request a Demo
- Content Title Description
QbD » Blog » State Of The Art Literature Review – Process and Documentation
Life Sciences Insights
Sharing expert knowledge via our latest blog posts
State Of The Art Literature Review – Process and Documentation
- Pia Gyselen, Lead Medical Writer at QbD Clinical
- July 3, 2023

A comprehensive, objective, and thorough systematic literature review to describe the general State of the Art and identify all relevant clinical safety, performance, and usability data of the device under evaluation (and/or equivalent device) should comply with strict regulatory guidelines as per EU MDR 2017/745 and MEDDEV 2.7/1 Rev. 4.
In this blog post, we would like to share with you the different steps and required documents for the execution of the systematic literature review.
FREE WHITE PAPER
State-of-the-art literature review.
In this white paper on Systematic Literature Review, we walk you through the different steps of conducting solid systematic SOTA literature searches.

State of the Art Literature Review – Process and Documentation
To plan and document the literature review searches and output, a well-designed and clearly written literature review plan or protocol (LRP) and report (LRR) are key and required for all device classifications, including all new and legacy devices and well-established technology (WET).
These documents are then also part of the technical documentation of a medical device. Both should be dated, version controlled, and signed by the regulatory writer, evaluator (preferably a clinical expert), and manufacturer.
Literature Review Plan
The literature review starts with the development of a literature review plan or protocol that should describe the background and scope of the literature review . This should include at a minimum:
- A rationale for the literature review;
- A description of the device, including the intended purpose, indications, target population, and users;
- The methods for identification, selection, and appraisal of the relevant publications to address the research questions, so the searches can be reproduced, data can be critically appraised, and the results can be verified.
Literature search strategy
The search strategy should be thorough and objective and identify all relevant favorable and unfavorable data related to the intended use, indication(s), target population, and performance of the device under evaluation and should cover:
- General SOTA, including clinical practice guidelines in the medical field
- Safety, performance, and usability data on similar benchmark devices/alternative treatments
- Safety, performance, and usability data on the device under evaluation and/or equivalent devices if equivalence is claimed
Different sources of clinical literature are applied. Multiple searches with different focuses, search criteria (with the correct use of Boolean logic), and filters (such as the publication type and date range) are required to obtain the necessary information and data.
MEDLINE (Pubmed) is the most commonly used search engine. Alternative databases are also designated such as but not limited to EMBASE, COCHRANE and Google Scholar.
For current practice guidelines in the respective medical field, more specific databases such as TRIP and UpToDate can be of interest. Justification for the selection of the respective databases should be provided.
To formulate the research questions to be answered by the literature review, unbiased, systematic, search methods should be used. A PICO-based search strategy is a generally accepted, evidence-based method.
After the generation of relevant keywords, the search string for the different searches has to be built. A rationale for the application of any in- and exclusion criteria, limits, and filters should be described in the plan.
Screening and selection process
Then the search output should be documented and tracked . A stepwise selection process should be applied. Selection of relevant literature should be objective and justification for exclusion of records properly documented.
Clinical safety, performance and usability data extracted from publications are subject to analysis and appraisal. According to Annex XIV of the EU MDR 2017/745 and MEDDEV 2.7/1 Rev. 4, all clinical data relevant to the device under evaluation should be appraised by evaluating their suitability for establishing the safety and performance of the device.
Literature Review Report
The output of the literature search and review is described and summarized in the literature review report (LRR). The body of the LRR consists of a description of the state of the art on the one hand and a systematic analysis of published clinical safety, performance, and usability data pertaining to the device and similar devices on the other hand. All data sets should be documented, adequately analyzed, appraised, summarised, and referenced in the LRR.
General SOTA
MEDDEV 2.7/1 rev. 4 appendix A9 provides guidance on the content of the SOTA section, i.e., the clinical background of the device under evaluation, the medical field(s) in which the device is being used, and the target population. Different alternative treatment options and similar competitor devices with (dis)advantages of each alternative should be discussed.
Under EU MDR 2017/745, increased scrutiny of alternative treatment methods for the same indication(s) is perceived. The benefits and risks of the device under evaluation and of similar benchmark devices and/or alternative treatments described in the literature should be detailed. This section also presents clinical practice guidelines and recommendations on the use of the device for the intended medical purpose. Finally, the general SOTA outlines unmet medical needs and the types of users of the device.
Clinical data on similar devices/alternative treatments
One of the objectives of the systematic literature review is to obtain the safety and performance acceptability criteria for the device under study . To this end, similar devices should be identified and listed upfront in the literature review plan at best, and data of similar competitor devices on the market should be extracted, analyzed and summarized in the literature review report.
In case no similar competitor devices are on the market or insufficient safety and performance data of competitor devices is publicly available, data of alternative SOTA treatments, preferentially devices, can be extracted for comparison to the device under evaluation in the clinical evaluation process. Alternative devices can for example concern devices with the same intended use, but different indication, patient population or mode of action.
Clinical data on device under evaluation/equivalent device
Last but not least, a search to retrieve safety, performance, and usability data of the device under evaluation (and equivalent device, if applicable) that is not generated by the manufacturer, should be set up.
A search for clinical literature related to the device under evaluation is typically done post-CE as part of the PMCF data collection in the CER. Obviously, to obtain these data, a separate search string containing the device trade name and manufacturer is compiled. Data extraction, appraisal, and analysis are performed in the same way as for similar competitor devices/alternative treatments.
Conclusion of the literature review
The literature review should draw firm conclusions with regard to the safety and performance of the device under evaluation and discuss this outcome in relation to similar benchmark devices or alternative treatment options.
Identify any gaps in the evidence to confirm compliance with GSPR under the normal conditions of the intended use of the device. Evaluate if the clinical literature supports the intended purpose as stated in the IFU and any benefit and clinical performance claims made for the device under evaluation.
How can we assist in consolidated State of the Art literature reviews?
The SOTA literature review, as part of the clinical evaluation process, is a time-consuming complex process that requires specific reading, writing, and strong analytical skills.
At QbD Clinical, we provide knowledge-based expertise to help you set out the clinical strategy and plan and document the clinical evaluation process throughout the journey of your medical device – from start to finish.
To speed up the preparation of the required documentation and ensure high quality deliverables, a team of regulatory affairs specialists, literature review experts, medical writers, medical advisors and project managers work closely together.
Take advantage of the multi-disciplinary expertise of QbD Clinical to speed up and deliver an MDR compliant SOTA literature review and clinical evaluation in an efficient way.
Our services at a glance:
- Gap analysis of SOTA literature review and clinical evaluation documents
- Systematic literature reviews – literature review plan and report
- Regulatory Medical Writing – CEP, CER, PMS plan and report, PMCF plan and report, PSUR
- Clinical medical writing – CIP and CIR
- Advise in clinical strategy
- Operational management of pre-and post-market clinical investigations and PMCF surveys
Did you find this article interesting? Thanks for sharing it with your network:
Read more experts content
- Regulatory Updates
- Whitepapers
Table of Contents
Stay up to date with life sciences insights.
- October 24-26
- Barcelona, Spain
Come visit our booth at CPHI Barcelona 2023
Come to see the QbD Group at stand #3G73 at CPHI Conference in Barcelona. And after the conference…Eat & Connect with lifescience professionals at our QbD’s CPHI Networking Drink .
- Lupe_Grau Cancel
The Clinical Evaluation Literature Search: 6 Tips to Save You Time and Stress
The literature search is a key part of the clinical evaluation. It usually involves numerous hours of work. This article will give you six tips to help you efficiently carry out and fully document the literature search.
In the literature search, manufacturers gather together scientific articles, among other reasons, to document the state of the art and provide evidence of the safety, performance, and clinical benefit of their device. But more on this later.
Tip 1: Use the information in the guidelines when searching for literature
There are now several MDCG documents available on the topic of the clinical evaluation, but none that describe how to carry out and document the literature search for the clinical evaluation.
a) MEDDEV 2.7/1 Revision 4 on the literature search
MEDDEV 2.7/1 is also the most important guideline for the literature search under the MDR. The Medical Device Coordination Group says the same.
“For general guidance on a literature search, see MEDDEV 2.7/1 Revision 4, A5. Literature search and literature review protocol, key elements”
Section D, MDCG 2020-13
Through the literature search, you find literature on the device under evaluation, the equivalent device and the state of the art, including alternative examination and treatment methods.
Annex 5 of MEDDEV 2.7/1 Revision 4 describes the most important aspects to remember when documenting the literature search. In it, the guideline requires the objective of the literature search(es) to be documented. Examples of such objectives are:
- Providing data on the device under evaluation (including device name and model)
- Identifying important data for the risk management process (focus on patient population and existing interventions)
- Providing information for the evaluation of the benefit/risk profile
- Giving an overview of the current safety specifications
- Enabling a comparison of possible side effects
- Providing information on benchmark devices
Manufacturers also have to document the search methods .
There will be more on documentation in the second tip.
Further information
Further information can be found in our in-depth article on MEDDEV 2.7/1.
b) MDCG documents
The MDCG documents do not currently offer any concrete guidance on how the literature search should be carried out. The MDCG document 2020-13 “Clinical evaluation assessment report template” is nevertheless useful:
It is primarily aimed at clinical evaluation reviewers, particularly notified bodies , but it also provides indirect guidance for anyone carrying out a clinical evaluation. Section D deals with literature search and literature review. The requirements listed in this section are the same as the ones in MEDDEV 2.7/1 Revision 4. The focus is on:
- Search categories (e.g. device search or state of the art including clinical condition)
- Scope of the search strategy
- Search and review methods
- Literature search documentation
The MDCG 2020-13 document refers to MEDDEV 2.7/1 Revision 4. So, save some time for the MDCG document and be glad that you can continue working with MEDDEV 2.7/1 Revision 4, especially when it comes to the literature search.
c) Other documents
Other guidance documents on the preparation of the clinical evaluation include, for example, IMDRF MDCE WG/N57FINAL:2019 .
Tip 2. Fully document the literature search
A) literature search protocol.
Incomplete documentation of the literature search will result in a non-conformity. This can lead to unnecessary queries or even deviations in the audit, since the MDCG document 2020-13 explicitly requires notified bodies to review the literature search documentation. It requires reviews to evaluate the following metadata:
- Search terms
- Databases used
- Inclusion and exclusion criteria
- Exclusion of duplicates
- Literature review procedure and documentation
- Search methods
These metadata are incorporated into the literature search protocol.
MDCG 2020-13 requires auditors to pay special attention to the exclusion criteria.
The clinical evaluation should clearly describe the selection criteria with respect to the regulatory purpose to which it will apply. The CER should clearly differentiate between the two types of data (device under evaluation or an equivalent device, state of the art or alternative treatment option). If the data does not relate to either of the above, provide a rationale with respect to its inclusion.
MDCG 2020-13 requires manufacturers to define and document the selection criteria for the literature searches. The selection criteria should be defined in the context of the clinical evaluation and distinguish between at least two searches for data or information:
- Search for the state of the art
- Search for the device under evaluation/equivalent device
You can find out more on these two searches in tip 3.
b) Additional documentation
The complete documentation of the literature search doesn’t just include the literature search protocol.
The documentation includes all the following documents:
- The aforementioned literature search protocol
- The literature search report, including any deviations from the literature search protocol and the results of the search
- Complete list of retrieved articles
- Complete list of articles excluded with reasons for exclusion
- Full text copies of relevant documents
Please note!
Most clinical evaluations that the Johner Institute receives for revision have been rejected by notified bodies because of literature searches where the above were not, or not completely, documented and available.
Tip 3: Remember that there are several literature searches
You are not free to choose what to look for. MEDDEV 2.7/1 Revision 4 requires your literature search to cover at least two essential topics:
- You need the state of the art search to demonstrate the state of the art for your device and to evaluate your device in comparison.
- You need the statements on your own device (or equivalent device) to demonstrate the safety, performance, and clinical benefit of your medical device.
Different search terms are used depending on the objective (see Figure 1).

You can’t neglect any of these searches. Otherwise there is a risk of a non-conformity in the audit.
The PICO method will help you with literature searches, for example, when trying to find search criteria for the state of the art.
P atient/ P opulation; I ntervention; C omparison; O utcome
The PICO method is used in evidence-based medicine in particular and recommended for use in clinical evaluation literature searches by MEDDEV 2.7/1 Revision 4.
Tip 4: Search in the databases relevant to you
Just as important as the search strategy is choosing which databases you are going to search. The MDR does not give any concrete advice on how to choose the literature databases. However, in article 2(48) it requires peer-reviewed publications.
With a few exceptions, PubMed only contains peer-reviewed publications. In contrast to Embase, PubMed allows free search and registration. However, access to the full texts is not always free of charge on PubMed.
In addition, the MDR mentions “relevant specialist literature” or “databases”. However, the EU regulation leaves it up to the authors of the clinical evaluation to decide which databases they search.
In section D, the MDCG 2020-13 states that multiple databases should be used to minimize bias in the literature review.
MEDDEV 2.7/1 Revision 4, Annex 4 provides some guidance for selecting suitable literature databases. It recommends using MEDLINE, PubMed and other databases such as EMBASE or the Cochrane CENTRAL trials register but does not explicitly require them.
Table 1: Recommended databases and possible benefits
So, you can save costs and start the search in PubMed and use additional databases (e.g., EMBASE) to cover European topics (therapies or medical devices in use in Europe).
You can find a list of other potential sources for literature and clinical data in the article on clinical data .
Tip 5: Use (Boolean) operators
The use of (Boolean) operators allows you to narrow down your literature search, which saves you the trouble of reading non-specific literature sources. However, you can also use Boolean operators to expand the search, especially if you don’t find enough literature sources.
The operators can be used to combine different search terms according to the context. The most well-known Boolean operators are AND, OR and NOT.
- Combining terms with "AND" filters the results for entries that contain all the search terms.
- Combining terms with “OR” filters the results to show entries that contain one of the search terms.
- And combing the terms with “NOT” excludes entries with this search term from your search.
Quotation marks and round brackets are also useful for improving the quality of the search results. Use them to get relevant and specific search results.
- If you put your search term in quotation marks, “”, the search engine will search for the search terms in that exact context and order.
- Round brackets, () can be used to refer a Boolean operator to terms or units.
The following table illustrates this with some examples:
Table 2: Use of Boolean operators
Note that the different databases use different operators. Check the database's website to find out more. This also affects the interpretation of the search phrase when no brackets are used, as shown in the last example in the previous table. Some databases interpret this search phrase as ("ice pack” AND “reduction of pain”) OR “reduction of edema”.
Tip 6: Don’t read the full text of all the search results
The literature search often returns several hundred publications. At this point, you may wonder whether it is necessary to read the full text of each publication.
You (still) don’t have to read the full texts of all the documents found in the initial search. You can exclude obviously non-relevant publications on the basis of the abstract if they are not related to the clinical evaluation and, for example, don’t have anything to do with the device under evaluation.
However, you should read the full text publication during the literature review at the latest. The MDCG agrees with this view:
“Abstracts lack sufficient detail to allow issues to be evaluated thoroughly and independently, but may be sufficient to allow a first evaluation of the relevance of a paper. Copies of the full text papers and documents should be obtained for the appraisal stage.”
Summary and conclusion
You should continue to use MEDDEV 2.7/1 Revision 4 as a guide for the literature search. The MDCG 2020-13 document refers to the requirements of MEDDEV 2.7/1 Revision 4 and encourages clinical evaluation reviewers to pay close attention to the following topics:
- Availability of full text publications
- Completeness of the meta-information (e.g., inclusion and exclusion criteria)
Control the range and specificity of the publications found in your literature search by using (Boolean) operators. Save time by excluding obviously irrelevant publications at an early stage, particularly if they do not relate to the state of the art or the device under evaluation.
Remember that in addition to the state of the art, you must at least research the information on equivalent devices. In other words, you must carry out at least two searches.
If you use the tips in this article, you will comply with the MDR’s requirements for literature searches.
Do you have any questions about implementation in specific cases? Email us or send us a contact request . We can review and trim down your documents to ensure conformity so that your clinical evaluation passes the audit.

Lea Wettlaufer

A quick overview: Our
Starter-Kit
Always up to date: Our
Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.
Individual Cookie Settings
Only accept required cookies.
Privacy Notes Imprint
Here is an overview of all cookies use
Required Cookies
These cookies are needed to let the basic page functionallity work correctly.
Show Cookie Informationen
Hide Cookie Information
Provide load balancing functionality.
Provides functions across pages.
Hubspot Forms
Used for the google recaptcha verification for online forms.
Cookies for Statistics
Statistic cookies anonymize your data and use it. These information will help us to learn, how the users are using our website.
Google Analytics
Tracking and analys of traffic on our websites.
Cookies for Marketing
Marketing cookies from thrid parties will be used to show personal advertisment. They use them to track users outside of their own web page.
Keeping track of a visitor's identity. It is passed to HubSpot on form submission and used when deduplicating contacts. It contains an opaque GUID to represent the current visitor. It also introduces cookies from linked in for marketing reasons.
LinkedIn conversion tracking.
Cookies for external Content
Content for Videoplatforms und Social Media Platforms will be disabled automaticly. To see content from external sources, you need to enable it in the cookie settings.
Google Maps
Used to display google maps on our Websites. Google uses cookies to identify and track users.
- Get The Newsletter!
- View The Latest Newsletter
- Follow me on Twitter
- Friend me on Facebook
- Connect with me on LinkedIn
- Subscribe to me on YouTube
- Subscribe to my RSS Feed
Quality/Regulatory

How Literature Review Automation Improves CER and PER Program Management

Beyond simply allowing for broader yet more efficient searches, leveraging software to automate literature reviews can organize references, assign screeners, and review screening decisions. This saves time, reduces bottlenecks, and, most importantly, leads to a highly transparent, standardized, and repeatable process that supports continuous CER and PER submissions across a product portfolio and for the life of a device.
Share this:
Manufacturers around the world are preparing for the upcoming implementation of the European Union’s Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) regulations. For medical device and diagnostics manufacturers, doing business in the EU is now subject to increased regulatory oversight. Among other challenges, this includes expanded requirements for demonstrating clinical evidence, defining state of the art technology , and conducting post-market surveillance and post-market clinical and performance follow-up.
With these additional requirements, state of the art program management for product portfolios takes on an increasingly vital role for manufacturers submitting a Clinical Evaluation Report (CER) for the MDR program or a Performance Evaluation Report (PER) for the IVDR counterpart.
Unfortunately, traditional CER and PER program management , which often includes ad hoc management, inconsistently applied processes across different product divisions, and the utilization of spreadsheets or non-purpose built software, are ill-equipped to meet more rigorous notified body submission expectations – including the frequency and breadth of literature reviews for multi-product portfolios that require annual submissions. In addition, literature reviews of the existing clinical evidence inform the need for post-market clinical and performance follow-up studies. This topic will be covered in subsequent business briefs in this series.
Compounding the lack of a standard, automated approach for literature views is the use of spreadsheets in their conduct. In fact, it is estimated that 90% of spreadsheets contain formula errors. These errors, coupled with the time and resources necessary to fix them, adversely impact the entire program management process. That leads to delays, omitted references, mistakes – or, worse, rejected submissions.
Implementing, however, can help firms take control of the program management process. Beyond simply allowing for broader yet more efficient searches, leveraging software to automate literature reviews can organize references, assign screeners, and review screening decisions. This saves time, reduces bottlenecks, and, most importantly, leads to a highly transparent, standardized, and repeatable process that supports continuous CER and PER submissions across a product portfolio and for the life of a device.
Understanding the Value of Literature Reviews
Before exploring how to automate literature reviews , it is important to first understand what a literature review entails. A literature review is a rigorous and robust method of evidence-based research used to collect, review, and report on large sets of published data in order to answer a well-defined research question.
Unlike a primary study, which collects data from an original source through methods such as a survey, randomized clinical trial, or case study, a literature review focuses on the entire catalog of data about a specific topic. Along with published scientific papers and studies, this data can include so-called gray literature such as white papers, unpublished manuscripts, and conference presentations.

The lifecycle of the literature review process consists of six key steps:
- Define the research question.
- Search relevant databases.
- Screen references for relevance.
- Full-text Retrieval.
- Extract and appraise the data’s quality.
- Document purpose, methods, results, and conclusions.
- Monitor for new content and adjust the research question if necessary.
All literature reviews aim to achieve the goals of efficiency, transparency, and reproducibility. Best practices such as dual independent screening, effective collaboration, wide-ranging searches, well-defined inclusion and exclusion criteria, and well-documented lists of excluded references are critical to achieving those goals.

Three Reasons Automated Literature Reviews Matter for Medical Device Companies
The literature review process is not without its challenges. Large datasets require significant effort and collaboration to be screened properly, but few research teams have the resources they need to support this process effectively.
Additionally, stringent MDR and IVDR requirements put more pressure on manufacturers to show transparent, systematic, and reproducible review processes; this increased workload is difficult to manage using ad hoc processes. This can lead to a rushed review that’s increasingly susceptible to human error and likely to be rejected by a notified body, which can result in lost revenue as a result of time-to-market delays.
Automating the literature review process, though, brings three important benefits to medical device companies preparing CER and PER submissions.
Do More Faster and Smarter
MDR and IVDR requirements force medical device manufacturers to increase the frequency, traceability, and overall documentation of clinical and product evaluation reports. This presents a number of specific challenges to research teams : most notably, a heavy documentation burden, ongoing monitoring for literature updates, continual updates to research libraries and other relevant project information, and an increased likelihood of user, bias, and conflict among reviews as the volume of literature increases.
All of these challenges lengthen the time necessary to complete a literature review. That is the last thing research teams need when reviews are both more frequent and more thorough. Automating the literature review process accelerates the screening of references, provides easier access to the source material, and immediately flags disagreements among reviewers that require resolution.
With automation in place , research teams can complete more accurate reviews in less time. For example, we work with one firm where automation has reduced review times by 30% as compared to reviews managed in an ad hoc manner with spreadsheets. Another firm has greatly reduced reference screening times for titles and abstracts by more than 70% and cut time for the creation of diagrams and flowcharts by more than 50%, all of which has translated into a seven-figure cost savings for the company.
Applying AI to Literature Reviews
Based in Canada, the Health Evidence Registry™ is a repository of over 6,500 critically appraised systematic reviews on the effectiveness of public health interventions maintained by the National Collaboration Center for Method and Tools (NCCMT), which is hosted by McMaster University in Hamilton, Ontario. Every month, they search and identify relevant systematic reviews for their repository. On average, their database searches return approximately 8,000-14,000 references monthly to screen.
With DistillerSR , NCCMT confidently reduced result sets by more than 75% with minimal false excludes. Working with a team of only 4-6 staff, the NCCMT team saves approximately 15-20 hours per month on screening. By reducing the time it takes to perform the initial screening, NCCMT can expedite their monthly processes and move relevant references on to critical appraisal and eventually upload them to the repository earlier. The approach makes it easier and faster to complete the entire monthly process.
“DistillerSR helped us create an easier workflow especially at the beginning of each month when the workload is heaviest,” said Maureen Dobbins, RN, PhD, Scientific Director, NCCMT, School of Nursing, McMaster University. “We’re at a point now, where doing it manually is no longer feasible and is a barrier to getting the research out to decision-makers ASAP.”
Take Control of the Submission Process
One manufacturer we worked with previously had a literature review process that was 100% manual. A medical writer would request articles from the library, obtain the abstracts, read and highlight the abstracts in a paper document, copy and paste these notes into a separate document, go back to the library to retrieve the full-text articles, and then set up inclusion and exclusion rules in a spreadsheet. This may be an extreme example, but many medical device manufacturers have similar processes in place. This type of approach poses numerous problems. It is unable to scale beyond a single submission; it won’t stand up to the scrutiny of the notified body; it varies based on the experience and industry knowledge of the individual medical writer.
To stand up to the requirements of CER and PER submissions , the literature review process needs to be configurable. Steps in this direction include review templates that adjust to meet the needs of a specific notified body, cloud-based project hosting that enables access for the entire team regardless of location, and seamless connections to corporate libraries. Manual reviews cannot support this level of configuration, but literature review software is positioned to do just that.
A configurable review process, then, is structured, standardized, and repeatable. It is capable of supporting continuous reviews for ongoing CER and PER submissions – some of which must be resubmitted annually for high-risk devices – and a broader device portfolio across the company. But the biggest benefit is time: Research teams spend less time completing logistical tasks such as data entry and more time using their expertise to complete a thorough review.
Manage a Single Source of Trusted Evidence
Spreadsheet-based processes lead to more than just inefficiency. Inconsistent manual processes for sharing, distributing, and tracking information increase the likelihood of errors, whether through incorrect formulas or copy-and-paste mistakes. In addition, these inconsistencies hinder gap analyses. With limited insight, gaps cannot be identified until the later stages of a review, which could require the revision of a research question. Altogether, this impacts the quality of the review – which can lead to rejected submissions and costly delays.Automation addresses these issues by allowing research teams to track all review activity. This provides real-time insight into the progress of the review as a whole, and it enables quality control measures such a s double-checking and verifying exclusions. The ability to capture and validate data as reviewers enter it , also provides valuable benefits, as it associates every action with the individual who affected it while removing the need for time-consuming data cleansing.
Ultimately, workflows are secure and contained within a single project, and the entire process is transparent to all participants – making it both defensible and auditable. A single source of truth also provides better insight into potential gaps and resourcing needs than an ad hoc, spreadsheet-driven process; this insight has the added benefit of creating a more informed and visible pipeline for reviews across a product portfolio or business unit.
Literature Review Automation Modernizes CER and PER Program Management
The most immediate impact of automated systematic reviews is improved efficiency, as research teams spend less time fixing preventable mistakes. However, the true impact extends further. Automated reviews enable a more transparent, repeatable, and auditable process, which allows manufacturers to create and implement a standard framework for literature reviews that can then be inserted into all CER and PER program management plans consistently across every product, division, and research groups.
With a repeatable process in place, program management can focus less on the nuance of literature reviews and more on critical management and communication tasks that help move a program along. In addition, a familiar process means fewer gaps and faster identification of the gaps that do exist, which reduces bottlenecks and delays. Standard workflows for all literature reviews, meanwhile, enable new team members to contribute proactively and quickly across portfolios, reducing training times and establishing a consistent CER and PER process. Finally, transparency and repeatability support a continuous submission process – which is critical for annual recertification of high-risk devices and ongoing program managing for products across multiple portfolios.
Traditional program management is ill-suited for the literature review process now mandated for ongoing compliance with the European Union’s MDR and IVDR regulations. Implementing automated literature reviews allows for faster and more accurate reviews while enabling the creation of a process-driven framework that can be applied to all future reviews. For manufacturers struggling to manage their pipeline of CER and PER submissions, an automated literature review process can bring much-needed order and set the stage for program management success.
Content sponsored by Evidence Partners .
Related Articles

Peter O’Blenis, CEO of Evidence Partners, discusses the growing role—and challenges –of literature reviews in…
The acquisition is expected to strengthen PTC’s closed-loop product lifecycle management offerings by extending the…

The Experiential Learning Program (ELP) provides CDRH review staff with an opportunity to visit sites…
The voluntary program is designed to help companies advance alternative and innovative ways to sterilize…
© Copyright 2015 - 2024 Innovative Publishing Co., Inc., All Rights Reserved
Other Innovative Publishing Co., Inc. Sites: Food Safety Tech | Cannabis Industry Journal

- Privacy Overview
- Strictly Necessary Cookies
- Cookie Policy
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
We use tracking pixels that set your arrival time at our website, this is used as part of our anti-spam and security measures. Disabling this tracking pixel would disable some of our security measures, and is therefore considered necessary for the safe operation of the website. This tracking pixel is cleared from your system when you delete files in your history.
We also use cookies to store your preferences regarding the setting of 3rd Party Cookies.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
A browser cookie is a small piece of data that is stored on your device to help websites and mobile apps remember things about you. Other technologies, including Web storage and identifiers associated with your device, may be used for similar purposes. In this policy, we say “cookies” to discuss all of these technologies.
Our Privacy Policy explains how we collect and use information from and about you when you use This website and certain other Innovative Publishing Co LLC services. This policy explains more about how we use cookies and your related choices.
How We Use Cookies
Data generated from cookies and other behavioral tracking technology is not made available to any outside parties, and is only used in the aggregate to make editorial decisions for the websites. Most browsers are initially set up to accept cookies, but you can reset your browser to refuse all cookies or to indicate when a cookie is being sent by visiting this Cookies Policy page. If your cookies are disabled in the browser, neither the tracking cookie nor the preference cookie is set, and you are in effect opted-out.
In other cases, our advertisers request to use third-party tracking to verify our ad delivery, or to remarket their products and/or services to you on other websites. You may opt-out of these tracking pixels by adjusting the Do Not Track settings in your browser, or by visiting the Network Advertising Initiative Opt Out page.
You have control over whether, how, and when cookies and other tracking technologies are installed on your devices. Although each browser is different, most browsers enable their users to access and edit their cookie preferences in their browser settings. The rejection or disabling of some cookies may impact certain features of the site or to cause some of the website’s services not to function properly.
Individuals may opt-out of 3rd Party Cookies used on IPC websites by adjusting your cookie preferences through this Cookie Preferences tool, or by setting web browser settings to refuse cookies and similar tracking mechanisms. Please note that web browsers operate using different identifiers. As such, you must adjust your settings in each web browser and for each computer or device on which you would like to opt-out on. Further, if you simply delete your cookies, you will need to remove cookies from your device after every visit to the websites. You may download a browser plugin that will help you maintain your opt-out choices by visiting www.aboutads.info/pmc. You may block cookies entirely by disabling cookie use in your browser or by setting your browser to ask for your permission before setting a cookie. Blocking cookies entirely may cause some websites to work incorrectly or less effectively.
The use of online tracking mechanisms by third parties is subject to those third parties’ own privacy policies, and not this Policy. If you prefer to prevent third parties from setting and accessing cookies on your computer, you may set your browser to block all cookies. Additionally, you may remove yourself from the targeted advertising of companies within the Network Advertising Initiative by opting out here , or of companies participating in the Digital Advertising Alliance program by opting out here .
Literature Search & Review for MDR Compliance
A guide to medical device literature searches & reviews as critical components in effective MDR compliance strategies
Guide topics
Looking for expert consulting support on your regulatory strategy? Contact us today for a free & confidential discussion about your options.
What is a medical device literature search & review?
A systematic literature search & review is a structured and objective process which identifies, critically appraises and analyses clinical evidence in order to answer carefully formulated research questions.
Literature Search Protocols & SOTA Reviews
Market-leading systematic literature reviews produced by clinically-active medical professionals.
Explore our literature search & review services
It involves a thorough, methodical search of databases such as PubMed and Google Scholar to identify all literature sources relevant to the clinical background (SOTA) and/ or the subject device.
A systematic literature search & review should be performed in line with a search protocol, setting out the search terms to be used, inclusion/ exclusion criteria, appraisal framework, analysis criteria and justifications for decisions. It should also identify requirements for the qualifications of the individuals assigned to the review.
Literature search & review forms an important component of MDR compliance , forming key aspects of Clinical Evaluation , Risk Management and other parts of all effective MDR regulatory strategies.
What is the role of literature search & review in achieving MDR compliance?
Literature search & review is a pivotal component of the Clinical Evaluation of medical devices under the MDR. The importance of a systematic review of available evidence through this process is emphasised repeatedly in MedDev 2.7/1 rev 4 and is crucial in ensuring compliance with the new EU MDR.
1. Clinical Evaluation

A literature search & review is a pivotal component of medical device Clinical Evaluation . According to MedDev 2.7/1 rev 4 , they are essential for identifying safety and/or performance data relating to both the clinical background (State-of-the-Art) and to the subject device itself.
The Clinical Evaluation Plan (CEP) should set out a carefully designed literature search & review protocol to guide conduct of the evaluation and ensure that objective, rigorous standards are adhered to.
Effective literature searches are even more important in the event of a claim of equivalence, since the results of literature review on the equivalent device will stand in place of a clinical investigation of the device under consideration.
It can be expected that the methodology and conduct of the literature search & review will be intensely scrutinised by Clinical Evaluation Report reviewers, so ensure you have the right advice and the right approach to this crucial aspect of Clinical Evaluation.
2. PMS & PMCF

As part of the Quality Management System (QMS) , manufacturers are expected to implement and maintain a Post-Market Surveillance (PMS) system that routinely evaluates the clinical performance and clinical safety of the device. The scope and nature of such PMS should correspond to the device and its intended use. PMS creates new data on a frequent basis (e.g. safety reports, results from published literature, registries, PMCF Studies, and other data about device usage). These data must be analysed for information that has the potential to alter the risk/benefit profile, as well as the clinical performance and clinical safety of the device.
Post-Market Clinical Follow-up (PMCF) activities should include scope for a regular, scheduled systematic review of the medical literature throughout the lifetime of a device; in turn, the results will inform the PMS Report/ PSUR, PMCF Report and the Clinical Evaluation Report (CER) .
3. To establish State-of-the-Art (SOTA)
A systematic review of the literature is essential for identifying and analysing sources of clinical data relating to the clinical background and State-of-the-Art. As per MedDev 2.7/1 rev 4:
The State-of-the-Art embodies what is currently and generally accepted as good practice. The most technologically advanced solution is not always the State-of-the-Art.
According to the MedDev guidelines, a literature review should be used to describe the clinical background and identify current knowledge/ State-of-the-Art in the corresponding medical field of the subject device.
Consideration of evidence relating to comparable alternatives to the subject device should enable the determination of safety and performance benchmarks against which the subject device may be compared in the Clinical Evaluation.
The applicability of a State-of-the-Art review is not restricted to Clinical Evaluation – it can also be vital in competitor analysis, market analysis, and in identifying opportunities for product improvement or innovation.
4. Risk Management
A literature search of independently published evidence also forms a key input into the Risk Management process. It’s an essential step in identifying hazards (including hazards due to substances, technologies, and manufacturing procedures) and in producing evidence-based estimates of the probability and severity of risk.
Regular literature reviews can help ensure that Risk Management remains current and up to date. relating to subject device and/ or equivalent device can contribute to an effective Risk Management process.
An ongoing literature search & review enables proactive Risk Management by allowing organisations to identify and escalate concerns as they arise or before they become apparent, allowing them to take a proactive and dynamic approach to Risk Management.
How to structure the literature review
- CER Writing Training Course
Implement a future-proof Clinical Evaluation process across your entire medical device portfolio.
Explore our CER Writing Training Course
According to MedDev 2.7/1 rev 4, a literature review should include the following information:
- Introduction to the subject device
- Objectives/purpose of the literature review
- Search protocol which includes inclusion/ exclusion criteria and an appraisal & analysis plan
- Research questions that set out what you intend to discover from the literature review
- Search terms
- Analysis and appraisal of the literature identified
- Safety and performance benchmarks identified from the analysis
- List of exclusions and justification
Each component should be properly planned and prepared to ensure that the document is understandable by a reviewer who may not be familiar with the medical device.
Useful resources
- Five useful resources when writing a medical device CER
Related guide topics
- Clinical Evaluation
- Clinical Evaluation Plan (CEP)
- Clinical Evaluation Report (CER)
- All Guide topics
- Share on LinkedIn
- Share on Facebook
- Share by email
Back to top
Do you need help with your regulatory strategy?
- Skip to main content
- Skip to FDA Search
- Skip to in this section menu
- Skip to footer links

The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
U.S. Food and Drug Administration
- Search
- Menu
- Medical Devices
- Science and Research | Medical Devices
Medical Device Material Safety Summaries
The U.S. Food and Drug Administration (FDA) partnered with ECRI (originally founded as Emergency Care Research Institute), an independent nonprofit organization, to perform a comprehensive literature search and systematic review to identify the current state of knowledge about medical device material performance after implantation.
As part of the FDA’s ongoing commitment to promote the safety of medical devices, the FDA and ECRI are publishing these safety summaries for materials that are commonly used in implantable medical devices and the effects of those materials on patients over time.
UPDATE February 2023: The FDA added 3 new safety summary reports to the list of safety summaries below:
- Colbalt Chromium (CoCr)
- Stainless Steel
The vast majority of patients implanted with medical devices have no adverse reactions. The device works and performs as expected to treat medical conditions or help patients better manage their health. However, a growing body of evidence suggests that a small number of patients may have biological responses to certain types of materials in implantable or insertable devices. For example, they develop inflammatory reactions and tissue changes causing pain and other symptoms that may interfere with their quality of life.
The FDA is also beginning to see manufacturers incorporate new types of materials in devices. CDRH’s Office of Science and Engineering Laboratories (OSEL) has been conducting a wide array of research studies to learn more about the new advances in device materials.
The FDA believes that an increased understanding of the behavior of materials over time will ultimately result in an overall improvement in the safety and effectiveness of medical devices.
Safety Summaries
The following safety summaries are now available. The FDA will continue to release reports as they become available.
- Acrylic acid derivatives , which includes di-, tri- and glycerol methacrylates, often used in dental resins
- Cobalt Chromium (CoCr)
- Cyanoacrylates
- Hyaluronic Acid (HA)
- PEG (polyethylene glycol) , used broadly and as a coating for stents and catheters
- PET (polyethylene terephthalate) , used broadly
- Poly (2-Hydroxyethyl Methacrylate) (pHEMA)
- Polycaprolactone (PCL)
- Polyether ether ketone (PEEK)
- Polyhydroxy acids, including PLA, PGA, and other blends and copolymers , the most common class of bioresorbable polymers
- Polymethyl methacrylate
- Polypropylene , often used in surgical mesh
- Polytetrafluoroethylene (PTFE)
- Polyurethanes
- Siloxanes , often used in breast implants
- Silver , used as an antimicrobial agent
Five Key Questions
In compiling this safety information, the team focused on the following five key questions about local and systemic response to materials commonly used in medical devices.
- What is the typical or expected local host response to the material?
- Does the material elicit a persistent or exaggerated response that may lead to systemic signs or symptoms – beyond known direct toxicity problems?
- Are there any patient-related factors that may predict, increase, or decrease the likelihood and/or severity of an exaggerated, sustained immunological/systemic response?
- Are there any material-related factors that may predict, increase, or decrease the likelihood and/or severity of an exaggerated, sustained immunological/systemic response?
- What critical information gaps exist and what research is needed to better understand this issue?
More Information
- CDRH/ECRI Project Presentation Video: 2021 Regulatory Education for Industry (REdI) Conference
- Statement from FDA Commissioner Scott Gottlieb, M.D. and Jeff Shuren, M.D., Director of the Center for Devices and Radiological Health, on efforts to evaluate materials in medical devices to address potential safety questions
- Biocompatibility Assessment Resource Center
- Safety of Metals and Other Materials Used in Medical Devices
Don't know where to start? Watch our free starter videos and save lots of time and consultant fees →
Articles Technical Documentation
Updated May 24, 2023
Clinical Evaluation: How to Write a Regulatory Compliant Literature Review

A clinical evaluation is required for all medical devices according to the MDR. The main task of a clinical evaluation is to identify pertinent data in relation to your software device and similar ones. This will help you to prove the intended use of your software device and the clinical claims, that your software device is safe, presents no risks or the benefits outweigh the risks, and ideally, the benefits outperform any existing devices, e.g., by comparing your software device with the existing ones (if any) or any studies from peer-reviewed publications, medical guidelines and reports. This is done through a systematic literature review and thus, better formalising an efficient process to avoid any potential pitfalls including notified body non-conformance.
If you want a brief overview of the Clinical Evaluation Report, check out our article Clinical Evaluation Report (CER) For Medical Devices: 3 Easy Steps . To understand what the notified bodies are looking for, our template Literature Evaluation Checklist summarises that. The common points of failure that we have heard about seem to be incomplete search coverage, incomplete audit trial, and data integrity/data errors.
A pragmatic literature review approach implies a simplified, repeatable, reproducible, transparent, reusable process. Below are some practical considerations to help you conduct a high-quality literature review to produce quality data output for your Clinical Evaluation Report.
Optimise your search terms strategy as early as possible
Unfortunately, you will be able to come up with a strategy for the terms to be used to screen databases through trial and error. Our approach starts by defining all possible MeSH terms that describe the intended use of your medical device and the intervention/therapy your medical device aims for. We then check for the most recent publications, e.g., a recently published systematic review and/or a meta-analysis, and write down the MeSH terms those authors used. We then define a few search queries and refine them if needed in order to cover the appraisal criteria and/or target systematic reviews and randomized controlled trials.
Most importantly, you will have to describe explicitly your search terms strategy, i.e., how you identified the relevant publications. If someone reads the Section Literature Search Protocol and follows it, the same list(s) of publications should be retrieved.
If you have the fear of not missing references or are willing to explore new fancy ways of deriving existing knowledge and have resources for that, you might want to consider natural language processing (models developed by Hugging Face, OpenAI) or network-derived tools (Inciteful, Open Knowledge Maps, to name a few). You might also want to consider checking the references for any similar medical device on the market.
Always capture reasons for the inclusion and exclusion of your literature
There is a big chance that auditors ask you why you excluded a certain study. Writing the inclusion and exclusion criteria in your Literature Search Protocol will ease not only the argumentation of the choices made but also, the evaluation process itself.
Some examples of inclusion criteria we follow:
- Type of studies, e.g., randomized controlled trials, systematic reviews/meta-analysis, observational studies (prospective/retrospective cohort studies, case-control/cross-sectional studies, case reports/case series/other non-analytic studies),
- The medical condition/disease,
- The intervention/method/digital therapy,
- Characteristics of patients/user profile, e.g., adults 18 years and older.
Some examples of exclusion criteria we follow:
- Duplicate references,
- Not available in English and German,
- Other medical areas,
- Animal studies.
Ensure using multiple data sources
Pulling data from multiple sources gives your report credibility in that all evidence was identified. On the other hand, this leads to additional work, mainly making sure of excluding duplications of references. One way we deduplicate our lists is by retrieving the PMIDs from the PubMed of all the searches. PubMed allows you to download the lists as a CSV file. We then use either a Python workflow or any other tools such as KNIME to identify and delete the duplicates. You can do this as well in Google Sheets by checking for conditional formatting based on any other variable and not necessarily the PMIDs, e.g., titles, list of authors etc.
Document all the references
You have to keep the lists of all your references somewhere. That includes both the relevant ones (plus their full text) and the ones you excluded from your analysis. Even though there are several tools (including free ones) to support you with the systematic review and specifically with this task, you might still want to consider Google Sheets (like us). One way to organise your Google Sheet is shown below. Feel free to adapt it to your needs.
Most importantly, no matter the tool you choose, make it a living document to add any new references and data sources. For example, you can set alerts in Google Scholar to help you with this.
Skim efficiently through the list to identify the relevant publications
We recommend using a two-step screening process. Firstly, review the title and abstract of your compiled list of publications for relevance following the appraisal criteria. Screen the full text of the identified and selected relevant publications for safety and performance data. This is time-consuming and you might even be in a position of paying to access publications. We don’t have a better solution for now. If you find one, let us know! Also, there might be consultants that would recommend you consider using dual screening, e.g., two people to screen the literature to avoid introducing errors and be confident in the findings. The roles are up to you. What matters is to describe how you went through the screening and document everything in e.g., a PRISMA diagram.
Evaluate and weigh your clinical data as a grading system
There are various methods used to appraise and weigh clinical data. The Appendix F of the IMDRF MDCE WG/N56 FINAL:2019 describes a grading system in two tables that we found to be pragmatic and recommend following it. Otherwise, you can define your own criteria and assessment method or rely on other existing approaches such as the ACC/AHA Recommendation System proposed by the American College of Cardiology (ACC) and the American Heart Association (AHA).
Summarise data effectively
The auditors will always appreciate a table over descriptions. Thus, turn unstructured content into structured and easy-to-follow content. Use tables whenever possible, e.g., for data comparison. Also, when you write about clinical data, summarise what your included studies showed, how does this relate to your medical device, which benefits those studies presented. Is your medical device expected to have the same benefits? Which risks were mentioned in those studies, and will those risks apply to your medical device? Thus, make sure to make the linkage between the outcomes you mention in the Clinical Evaluation Plan and how the outcomes were evaluated in the identified studies and what benefits, risks and performance were concluded.
Lastly, if you are using our template for your Clinical Evaluation Report, there are three Sections which you should consider focusing your time on mostly: Clinical Background, Current Knowledge, State of the Art, Section Literature Search, and Section Clinical Data. We recommend starting with writing the Clinical Background, Current Knowledge, State of the Art, especially for situations when there is a clear medical indication. Then, continue with Literature Search followed by the Clinical Data, and finalise the rest of the document. For the identified clinical studies in the scientific literature, consider assessing them for all criteria at once to avoid redundancy in reading a publication multiple times: device names assessed by the authors, relevance based on the literature appraisal criteria, level of the evidence, tendency, comparability, performance, safety, clinical information such as patients, study design, measured outcomes. You can organise the information in tables. Also, I would write the Clinical Evaluation Plan firstly but not try to have a complete version of it done immediately. I would rather come back to refine it after I’m done with the Clinical Evaluation Report. This is because I will know exactly how I identified the relevant publications, the potential safety issues based on those publications, and performance claims.
On a slighty different note: You want to get your medical software certified under MDR but don't know where to start? No worries! That's why we built the Wizard . It's a self-guided video course which helps you create your documentation yourself. No prior knowledge required. You should check it out .
Or, if you're looking for the most awesome (in our opinion) eQMS software to manage your documentation, look no further. We've built Formwork , and it even has a free version!
If you're looking for human help, did you know that we also provide some limited consulting ? It's limited because we are not many people. We guide startups from start to finish in their medical device compliance.
Congratulations! You read this far.
Get notified when we post something new.
Sign up for our free newsletter.
I am a scientist with a background in Pharmaceutical Sciences and computational modelling passionate about digital health technologies. I joined OpenRegulatory for the shared vision and values. I dream of a world where each patient has equal access to effective and safer AI solutions. Let’s make it happen together!
If you have any questions or would like to share your opinion publicly, feel free to comment below. If you'd like to reach out privately, send us a message .
- US: 1.800.472.6477
- EU: +353 21 212 8530
QA/RA Consulting, Auditing & Training
Let's get started
- November 2, 2023
Does Your Medical Device CER Meet EU MDR Requirements? Creating a Solid Clinical Evaluation Process

In this guide:
Stages in the clinical evaluation process Defining the scope and drafting a plan Making sure CER evaluators are qualified Creating a literature review protocol Identifying data needed to fulfill plan requirements Crafting a literature search and review strategy Choosing the appropriate data Is your data valid and relevant? Appraising the clinical data Analyzing your datasets and drawing conclusions Clinical data analysis Alignment between clinical evaluation, IFU, and risk management Are additional clinical investigations needed? Writing your CER and when to update it Compiling the clinical evaluation report Creating your EU CER template Create a CER checklist How often your CER should be updated
Medical device regulatory professionals have been grappling with tighter requirements for clinical data to support clinical evidence since MEDDEV 2.7/1 Rev. 4 was released in mid-2016. Now that the European Medical Device Regulation (2017/745) is nearing implementation, clinical evaluation reports (CERs) have taken on new urgency.
Regardless of whether you are making updates to existing CERs or building one for a product launch, you want to make sure that you don’t experience any nasty surprises during your Notified Body audit and technical documentation review. The first step is to create a robust clinical evaluation plan.
As you probably know, medical device manufacturers can fulfill clinical evidence requirements using one or more of the following:
- Premarket clinical studies (the mostoption, but necessary for new technology/indications)
- Literature searches for existing clinical studies for equivalent devices (
- Compliance with harmonized standards (e.g., IEC 60601-1 on electrical safety)
- Data generated from PMS activities and postmarket clinical studies, such as postmarket clinical follow-up (PMCF)
- Clinical investigations
- Postmarket clinical follow-up
Stages in the Medical Device Clinical Evaluation Process
Before we go into detail on the process, here’s an overview of the various stages outlined in MEDDEV 2.7/1 Rev. 4 and where you will find guidance on each stage.

Clinical Evaluation Stage 0: Defining the Scope and Drafting a Clinical Evaluation Plan
Published literature is a key component of the clinical data gathered by most companies. While it may be tempting to dig in and start doing literature searches straight away, it’s important that you understand what is needed in the first place. This “scoping” process is vital because you will need to explain and defend it to your Notified Body, and you will need to replicate it in the future.
Your clinical evaluation plan will define the extent of information gathered based on the General Safety and Performance Requirements in Annex I of the MDR. MEDDEV 2.7/1 Rev. 4 defines numerous aspects that should be considered in a thorough clinical evaluation plan. You can find much more detail on the elements to be considered during Stage 0 in Section 7 of the MEDDEV, which delineates between new medical devices and those that already have CE Marking.
MEDDEV 2.7/1 Rev. 4 states : Clinical evaluation is necessary and important because it ensures that the evaluation of safety and performance of the device is based on sufficient clinical evidence throughout the lifetime that the medical device is on the market.
Here are some of the aspects that need to be included in your CER scoping process. Some of these issues apply only to “new” devices getting CE Marking for the first time.
- Device description – see Appendix A3 of the MEDDEV
- CE Marking status of the device
- Where the device is marketed outside Europe
- Design features, names, models, sizes, components, etc.
- Intended use of the device and special indications or target populations
- Risk management documentation
- Current device State Of The Art
- Data sources and types to be used
- Information needed for demonstrating Equivalence (new devices only)
- Design, manufacturing, materials, or labeling changes (devices with CE Marking)
- Any newly emerged clinical concerns (devices with CE Marking)
- New data generated from PMS (devices with CE Marking)
Of course, the amount of data deemed necessary to meet sufficient clinical evidence and the General Safety and Performance Requirements will also be determined by the nature of the device, its stage in the life cycle, and its safety record.
Article 1(a) of Annex XIV in the MDR provides additional detail about what the clinical evaluation plan should include. You’ll want to study this in addition to Section 7 of the MEDDEV before crafting your plan.
Making Sure CER Evaluators Are Qualified
Before you dive into planning, you’ll also want to know that the MEDDEV sets some pretty specific guidelines for who can take on this important task. Evaluating clinical data is serious business, and regulators want to ensure that the people performing the evaluation are well qualified to do so. Section 6.4 of the MEDDEV outlines the basic requirements. Evaluators should have an appropriate college degree plus 5 years of professional experience or 10 years of experience if a degree is not a prerequisite. In addition, Section 6.4 goes on to say that evaluators should have knowledge of:
- Research methodology
- Information management
- Regulatory requirements
- Medical writing
- Application of the specific device technology
- Diagnosis and management of the conditions to be diagnosed or managed by the device
These qualifications will need to be documented and you can be certain that your Notified Body will review documentation related to reviewer qualifications.
Creating an EU CER Literature Review Protocol and Reviewing Medical Device Clinical Data
Once you have created a robust clinical evaluation plan for your medical device(s), it’s time to get busy figuring out what data you need, drafting a protocol, and then taking stock of that data. This is considered Stages 1 and 2 of the CER process and the specifics can be found in MEDDEV 2.7/1 rev. 4, as shown below.
Clinical Evaluation Stage 1: Identifying Data Needed to Fulfill the Requirements of Your Plan

There are two broad categories of clinical data.
1 – Data not generated by your company (retrieved from online literature searches and other offline data)
2 – Data generated by your company, which can include:
- Premarket clinical investigations, including bench testing reports
- Postmarket data gathered from risk management and PMS activities, including PMCF studies, device registries, and more
Crafting a CER Literature Search and Review Strategy
For many companies, the data retrieved from literature searches will represent most, if not all, of the data they collect. That’s why it is so critically important that you develop a literature search strategy that is robust and can be replicated during subsequent updates to your CER. The output of your search and review should obviously include literature on your device and any identified equivalent device , plus a review of the current state of the art. Your literature search protocol should include the following elements (for more specifics on this, see Appendix A5 in MEDDEV 2.7/1 Rev. 4):
- Sources of data you will use (e.g., MEDLINE/PubMed, Embase, Google Scholar, ResearchGate, internet searches, etc.)
- The methodology you plan to use for searches
- Exact search terms and parameters (e.g., dates) used to search scientific databases and the internet
- Your specific selection or exclusion criteria along with justification for each
- How you will address duplication of data from multiple sources
- How you will ensure data integrity (e.g., QC methods or second reviewers)
- How you will appraise each data source and its relevance to your device
- How you will go about analyzing and processing the data
You should think of your plan as you would a standard operating procedure (SOP) or a detailed instruction. As an example, if you were to leave the company a year from now, your successor should be able to read your protocol and understand exactly what was done during the previous update.
Make sure you treat the literature searches related to device equivalence as a separate activity from the searches on current state of the art. Separately define and track search terms used, database sources, date parameters, etc. Also, be cognizant that your idea of “state of the art” may differ from reality – here’s why . Before finalizing your protocol, test it. Identify known papers relevant to your device or current state of the art and then test those search parameters to make sure those known papers appear in your search results.
Review It All: The Favorable…and the Not So Favorable
Your literature searches should be extremely thorough and encompass a breadth of search criteria. They also need to be documented in detail so the results can be independently verified and replicated. Your selection of literature should be objective (the good and the bad) and justifiable. Nearly every published study has citations that may lead you to additional relevant data. Again, this is where planning and staying within your device search parameters become really important, because you can very quickly wander down a virtual rabbit hole and not be able to replicate how you got there.
While most publications are available in English (e.g., 93% on MEDLINE), make some attempts to do basic searches in French, German, or Japanese using online translation tools. Obviously, if you work for a larger company that has offices in Europe or Asia, getting help from native-speaking colleagues would be ideal. While the odds are good that you will find what you need in English, you might uncover valuable data that can be translated and further appraised if needed. Also, if you are updating an existing CER, we recommend that you start your search a few weeks before the end date of previous search to ensure you don’t miss anything that may have been added at the very end of your previous search period but had not yet been indexed online.
Keep in mind that some data may be publicly available but not easily accessible online. For instance, the label and IFU of a competitive device (if using one) may yield useful information. Also, presentations made at industry conferences may provide information that is not available (or has not yet been published) online.
Clinical Evaluation Stage 2: Choosing the Appropriate Data Sets Also Requires a Plan
During Stage 0, you came up with a clinical evaluation plan focused on determining which data you need and from what sources. In Stage 2, your focus will turn to choosing the right data among the data sets you have found. This requires yet more planning. Your data appraisal plan should address:
- Criteria for determining the quality, relevance, and validity of the data
- How you will weigh the data related to favorable and unfavorable results
According to Section 9.2 of MEDDEV 2.7/1 Rev. 4, the criteria you select should “reflect the nature, history and intended clinical use of the device.” This is something you must document and justify based on current state of the art.
Is Your Data Valid and Relevant?
So you found plenty of data in Stage 1 that seem relevant to your device and its intended purpose. But how do you know if the quality is good? At first glance, you don’t. Sure, a paper published in The Lancet is instantly credible, but the quality of a publication cannot serve as the sole rationale for selection. Ultimately you need to dive headfirst into the pool and do investigative research of your own to appraise the quality of the data.
Doing this requires some skill, because it is on you to evaluate the methodology used to collect the data and therefore determine its scientific merit. As part of this appraisal process you will also need to weigh the contribution of each data set to the overall clinical evaluation. Section 9.3 of the MEDDEV contains seven pages of advice on how to evaluate the methodological quality and scientific validity of data sets.
A Higher Bar for Medical Device Equivalency Under the MDR
Many companies use equivalency claims to avoid having to conduct redundant pre- or postmarket clinical studies that prove safety and performance. This part of the clinical evaluation process is not new. What is new is the level of scrutiny those comparative evaluations will endure. The clinical evaluation requirements in the existing Medical Devices Directive (93/42/EEC) and MEDDEV 2.7/1 Rev. 3 largely favored device equivalency but did not define it. Thus, some companies took a liberal view of “equivalent.” When MEDDEV 2.7/1 Rev. 4 was released in mid-2016, it gave manufacturers a lot less latitude for determining which devices could be considered equivalent. In fact, Appendix A1 in Rev. 4 is quite clear about the clinical, technical, and biological characteristics your device must have in common with an “equivalent” device.
The MDR furthers tightens the screws for Class III and implantable devices, requiring a more in-depth assessment and making it more challenging to leverage competitor data for new devices. That’s because Article 61, Section 5 of the MDR requires manufacturers of such devices to have access to the full technical documentation of the competitive device(s). It also instructs Notified Bodies to ask for proof that you have a contract in place granting you permanent access to that technical documentation. Good luck with that.
Appraising the Medical Device Clinical Data
With an appraisal plan created and a grip on how to execute it, you can begin the hard work of appraising the data you have found. This appraisal must be done based on the complete text of publications you find, not just by reading the abstracts or summaries. For each document you appraise, you are required to document your appraisal of it to the point that it could reasonably be reviewed by others. The appraisal results should also support conclusions you are making about clinical safety and clinical performance of the finished device (e.g., citing non-device-related literature would be ranked low for appraisal).
Appendix A6 in MEDDEV 2.7/1 Rev. 4 can be helpful in performing your appraisal of data. It provides some examples of red flags that should make you pause, including clinical data that:
- Lacks basic information such as the methods used, number of patients, identity of products, etc.
- Has data sets that are too small to be statistically significant
- Contains data that applies improper statistical methods
- Employs studies that lack adequate controls
- Has an improper collection of mortality and serious adverse event data
- Depicts a misrepresentation by the authors
The issue of evaluating statistical methods and significance intimidates many RA/QA professionals. If you feel uncomfortable about your “stats skills” for conducting statistical analysis or reviewing statistical information, hire some outside expertise to evaluate these specific aspects. Better to invest in doing that now rather than have a Notified Body reviewer challenge your appraisal later.
Analyzing Your Medical Device Clinical Datasets and Drawing Conclusions
Anyone who has ever researched and compiled an entire European clinical evaluation report knows that the devil is in the details. Finding appropriate clinical data is not necessarily the most challenging aspect of the process – it’s the appraisal and analysis of that data that causes regulatory heartburn.
Many professionals interchange appraisal and analysis, but those are actually two distinct steps in the process. When you appraise data, you are looking to make sure it has statistically significant data sets, uses proper statistical methods, has adequate controls, and properly collects mortality and/or serious adverse event data.
During the analysis stage (Stage 3) you will really dig in and conduct a comprehensive assessment to determine if the data you have found meets clinical safety requirements, clinical performance requirements, and General Safety and Performance Requirements (GSPR) of the EU Medical Device Regulation (MDR). You will evaluate the following:
- Is the performance bench testing
- Is the risk-benefit ratio appropriate based on the intended purpose of the device?
- Can the device actually achieve all performance claims made by the manufacturer?
- Are the materials (e.g., IFU) supplied by the manufacturer adequate to describe the intended purpose and mitigate risk?
Clinical Evaluation Stage 3: Clinical Data Analysis

During Stage 3 you are expected to analyze the data to ensure the clinical evaluation demonstrates that any risks are minimal and acceptable. You also need to consider all aspects of the device’s intended purpose. You’ll find more detail on this in Section 10.2 of MEDDEV 2.7/1 Rev. 4, and in Annex AVII. You will identify gaps related to:
- Understanding the interaction between the device and body
- The completeness of the data available
- Type and adequacy of patient monitoring
- Number and severity of adverse events
- Severity and history of the condition being treated or diagnosed
- Current standards of care
- Other factors
Data from the literature you have appraised is often put into Excel tables to be analyzed. It’s a convenient way to compare different study details, patient populations, endpoints, adverse events, etc. This can end up being a sizable amount of data unto itself, but certain aspects can be parsed into “bite-sized” tables focused on particular issues. While spreadsheets are not especially wonderful for narrative explanations, they are useful as quick comparative tools and are extremely helpful in noting differences between studies when writing the summary.
Alignment Between Clinical Evaluation, IFU, and Risk Management
Your analysis should also examine the alignment between the clinical evaluation, labeling/instructions for use (IFU), and the risk management file, as well as the current state of the art . Reviewers need to pay very close attention to make sure that, for example, the IFU and promotional materials are harmonized with regard to medical conditions and target populations. This analysis also needs to be consistent with the appraisal you conducted during Stage 2. Here’s what Appendix A7.1 of MEDDEV 2.7/1 Rev. 4 has to say about it:
“The information materials supplied by the manufacturer (including label, IFU, available promotional materials including accompanying documents possibly foreseen by the manufacturer), should be reviewed to ensure they are consistent with the relevant clinical data appraised in Stage 2 and that all the hazards, information on risk mitigation, and other clinically relevant information have been identified appropriately.”
It is expected that you will conduct the analysis with input from your risk management files and appropriate standards such as IEC 60601-1 (electrical safety) and EN 62366 (usability). The goal, of course, is to demonstrate that any risks associated with the intended purpose of your device are acceptable when weighed against the benefits it offers the patient or user.
Are Additional Clinical Investigations Needed?
How much is enough? There is no definitive rule that determines whether you have collected enough clinical data to meet the General Safety and Performance Requirements (GSPR) of the EU Medical Device Regulation (MDR). You need to conduct a detailed gap analysis and come to your own conclusion about whether supplemental clinical investigations will be required. At this time, you should also determine whether there are residual risks and uncertainties. This might include factors related to rare complications, long-term performance, or safety under widespread use. You’ll find more information on this in Appendix A2 of MEDDEV 2.7/1 Rev. 4.
Writing Your EU MDR Clinical Evaluation Report and When to Update It
You’ve done a lot of work to get this far. Writing a medical device clinical evaluation report (CER) is the culmination of a monumental effort to conduct literature searches, find/review literature, and/or conduct original clinical investigations. Data must be sourced, appraised, analyzed, and then summarized into your CER. This final process – writing the CER itself – is commonly referred to as Stage 4.
Clinical Evaluation Stage 4: Compiling the CER
Creating Your EU CER Template
Because the contents of a clinical evaluation report vary according to the nature and history of the device being evaluated, neither MEDDEV 2.7/1 Rev. 4 nor the EU MDR provide a detailed CER template. However, Appendix A9 of the MEDDEV does provide nearly six pages of guidance on what the structure of your CER should look like and what content it should contain. Here’s the basic outline.
1 – Summary
2 – Scope of the clinical evaluation
3 – Clinical background, current knowledge, and state of the art
4 – Device under evaluation:
- Type of evaluation
- Demonstration of equivalence, if applicable
- Clinical data generated and held by manufacturer
- Clinical data generated from literature searches
- Summary and appraisal of clinical data
- Analysis of the clinical data
5 – Conclusions
6 – Date of next clinical evaluation
7 – Dates and signatures
8 – Qualification of the responsible evaluators
9 – References
Create a CER Checklist: Do You Have Everything Covered?
You’ve done all the hard work, but are you sure you didn’t forget anything? The checklist found in Appendix A10 should help. Here’s an abbreviated version of it. (Note that this is only a partial list, to give you a flavor of what your CER checklist should include.)
- Is the report understandable to a third party and does it provide sufficient detail?
- Is all data generated, mentioned, and summarized in the report?
- If claiming equivalence, are differences adequately disclosed and explained as to why you don’t expect them to affect safety and performance?
- Has the latest PMS/PMCF data been taken into account and summarized?
- Is current state of the art explained and substantiated?
- Are undesirable side effects and the risk/benefit profile acceptable when compared to state of the art?
- Is conformity to MDR General Safety and Performance Requirements stated?
- Do informational materials supplied correspond with contents of the report?
- Does the report identify all residual risks or uncertainties that should be addressed in PMS/PMCF studies?
- Is the report dated and qualification of the evaluators included?
If the CER covers several models, sizes, settings, or situations, are the conclusions correct for:
- All devices, sizes, models, and settings?
- Every medical indication?
- The entire target population and all intended users?
- The duration of product use, including repeat exposure?
How Often Your EU CER (Clinical Evaluation Reports) Should Be Updated
Section 6.2.3 of MEDDEV 2.7/1 Rev. 4 provides guidance to manufacturers on how often to update clinical evaluations. It says that the “manufacturer should define and justify the frequency” of CER updates. Typically, this is done in concert with your Notified Body audit and certificate renewal, but that predefined schedule can be tossed out the window if your postmarket surveillance activities uncover new risks.
Shown below is an example of how you could rationalize the frequency of updates to your CER. This sample shows a simple evaluation table for an ECG machine that is well established on the market and has decent clinical history. You can, of course, create your own rating system with more factors, and you could weight these factors as well. We’ve kept the table simple in order to illustrate the point that your NB wants to see that you have a systematic approach to determining your CER update schedule.

As you can see in this example, even though this device is an established ECG with good clinical history, due to its inherently complexity, where it is used on the body, and the target population on which it is used, updating the CER every few years is still recommended.
Regardless of your proposed rationalization for the update schedule, you must update the CER after getting new information from your postmarket surveillance activities that might change your current evaluation. For example, safety reports, newly published literature, or PMCF studies may uncover previously unknown safety concerns. Data from these sources must be to evaluated because it may change your risk/benefit profile. Remember that Annex XIV, Part B of the MDR mandates that your PMCF plan specify the “methods and procedures for proactively collecting and evaluating clinical data.”
Your CER Is a Critical Component of Your Technical Documentation
Along with your risk management documentation and postmarket surveillance plan, the clinical evaluation report is a centerpiece of the technical documentation needed for CE Marking. While we think of the CER primarily as a regulatory exercise, it’s important to remember that the ultimate goal is the advancement of patient safety. Looking at it through that lens will help you focus on the right things and create a CER in full compliance with MEDDEV 2.7/1 Rev. 4 and the EU MDR (2017/745).
Advance Your Knowledge of Clinical Evaluation Reports
If you found this article to be informative and you want to take the next step in advancing your knowledge of all things CER, consider our EU CER training class . Our consultants are also available to help you with EU CER development and gap analysis .
Our team is here to help. Contact us online
Get answers right now. call.

US Office Washington DC
1.800.472.6477
EU Office Cork, Ireland

+353 21 212 8530

Search by Topic
- All Category
- CAPA and Root Cause Analysis
- Clinical Evaluation Reports
- Combination Products
- Complaint Handling and PMS
- Design Control
- EU IVDR (2017/746)
- EU MDR (2017/745)
- FDA 21 CFR Part 820
- FDA 510(k) Submissions
- ISO 13485 QMS Compliance
- ISO 14971 Risk Management
- Lab Developed Tests
- Pharmaceuticals
- QMS Auditing
- Software and Cybersecurity
- Supplier Management
- Uncategorized

UNITED STATES 1055 Thomas Jefferson St. NW Suite 304 Washington, DC 20007 Phone: 1.800.472.6477
4 Emmet House, Barrack Square Ballincollig Cork, Ireland Phone: +353 21 212 8530
- Consulting & Auditing
Find a Training Course
- International Offices
Consulting & Auditing
- Medical Device RA/QA
- MDSAP, ISO 13845, QSR, & EU MDR
- Pharmaceutical RA/QA
- Connected Medical Technology
- Performance Excellence
- ISO 9000 & Related Standards
© Oriel STAT A MATRIX. All Rights Reserved.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- J Healthc Eng
- v.2023; 2023
- PMC10241585
Strategies for Medical Device Development: User and Stakeholder Perceptions
I-ching tsai.
1 Department of Biomedical Engineering, National Cheng Kung University, No. 1, University Road, Tainan City 701, Taiwan
Ching-Da Wang
Peng-ting chen.
2 International Institute of Medical Device Innovation, National Cheng Kung University, No. 1, University Road, Tainan City 701, Taiwan
Associated Data
The data used in this article cannot be shared publicly due to privacy reasons of the participants of the study.
Medical device development involves user safety, and it is governed by specific regulations. The failure of medical device developers to consider the influence of users, the environment, and related organizations on product development during the design and development process can result in added risks to the use of medical technologies. Although many studies have examined the medical device development process, there has been no systematic and comprehensive assessment of the key factors affecting medical device development. This research synthesized the value of medical device industry stakeholders' experiences through a literature review and interviews with industry experts. Then, it establishes an FIA-NRM model to identify the key factors affecting medical device development and suggests appropriate pathways for improvement. Results indicate that the development of medical devices should begin with stabilizing organizational characteristics, followed by strengthening technical capability and use environment, and finally, consideration should be given to the user action of medical devices. The results provide medical device developers with optimal development pathways and resource allocation recommendations to support developers in developing medical device development strategies as well as ensuring the safety and effectiveness of the products for end users.
1. Introduction
Medical devices have brought numerous benefits and contributions to human health; however, regulations and medical particularities increase the costs and sensitivity of medical device research, design, and clinical applications [ 1 ]. These situations pose considerable challenges to the developers, especially small-to-medium enterprises (SMEs) with limited operational resources [ 2 , 3 ]. When the medical device industry fails to evaluate numerous medical device designs for the usability in the product development process, it can lead to various problems in their applicability and can give rise to risks owing to the insufficient consideration of human, environmental, and organizational factors [ 4 , 5 ]. Therefore, medical device developers need to be aware of the various aspects of the development process and focus on human factors and compliance with medical device marketing regulations to reduce postmarket risks. However, an important research gap exists regarding how to invest limited resources in product development and how to formulate development strategies to achieve optimal healthcare benefits based on product efficiency, medical regulations, and user needs [ 6 ].
Many studies have examined various key factors in the medical device development process, including user operation [ 7 ], risks [ 8 ], effectiveness and regulation [ 9 ], and public stereotypes [ 10 , 11 ]. Although research in medical device development continues to grow in the field of medical engineering, there is still a lack of a more systematic and comprehensive research framework based on stakeholder perspectives to evaluate the crucial elements of the medical device development process [ 12 ]. There is a need for empirical accounts of medical device development factors as perceived by stakeholders. This study analyzed the key factors in the medical device development process using stakeholder interviews and questionnaires. The results from this study can help medical device developers to prioritize and allocate resources to critical items in the medical device development process.
2. Literature Review
2.1. medical device innovation challenge.
The growing demand for healthcare has made medical devices increasingly important in the healthcare industry. Countries classify medical devices according to their associated “user risk” as a basis for quality and safety management, regulatory control, or market licensing. Owing to the particularity of the medical industry and devices, governments around the world have clear classifications and strict specifications for the development and marketing of medical devices. Such regulations make the design, development, and market planning of this industry more challenging than those of general commodities [ 6 ]. One of the problems of medical device usage involves the medical device developer's failure to consider fully the application status of a product during the product design and development process, thereby resulting in equipment inapplicability. This problem may be due to a mismatch between user characteristics, product interface, and functions or the usage environment, which may affect users' cognition or cause operation errors, thereby leading to risks and injuries [ 13 ]. Overall, the ability to effectively integrate input from developers, organizations, and users in the early stages of medical device development to evaluate product design, confirm usability and save costs to develop safe and effective medical device products will provide sustainable benefits to developers and users.
2.2. Medical Device Development Considerations
The main purpose of a medical device is to meet indications for use and user needs. Medical device developers are often unable to understand the benefits of focusing on human engineering in the development of medical devices due to their inability to implement user involvement in the design process [ 14 ]. Therefore, developers must provide product education and training to improve operation skills and increase user confidence and trust in products [ 15 ]. In addition to incorporating human behaviour, capabilities, and limitations into the design of medical product systems, it is crucial to take into account individual user differences [ 16 ]. This includes technical knowledge, experience, and education.
Medical devices can be used in clinical or nonclinical settings, such as community homes and public settings [ 17 ]. Numerous factors related to the environment, organizational characteristics, and user status can affect the use of a medical device [ 18 ]. In the development process, the characteristics of an intended use environment (e.g., time, pressure, lighting, noise, temperature, and physical layout) can help developers understand the operation of a product and can optimize the use efficiency [ 19 , 20 ] as well as improve safety and effectiveness. The development of medical devices requires consideration of the opinions of different stakeholders, the management, and the culture that can be critical to the success of the product. A variety of factors can influence the product development strategy of medical device developers, including cost control, professional manpower, budget availability, and performance expectations [ 21 ]. Given the particularity of medical devices, market strategies should consider social backgrounds, reimbursement processes, and regulatory policies [ 9 ].
This study identifies four dimensions of medical device development, including user action (UA), technical capability (TC), use environment (UE), and organizational characteristic (OC), to comprehensively assess the key factors of medical device development as a piece of advice for medical device development and medical industry.
3. Research Design
After conducting a literature review and examining case studies of medical device development, this study gathered specific information that influenced medical device development. For the professional experience data collection, semistructured interviews with key stakeholders were used. Key stakeholders include two managers of auditing organisations, a CEO of a consultant company, two managers of a government agency, and a product manager of a medical device company. The interview included open-ended questions, focusing on the process of medical device development and key factors. From the interviews, key aspects and factors related to medical device development were extracted and used to design questionnaires. The questionnaires were distributed among experts and their responses were recorded. The DEMATEL method was employed to analyze the questionnaire data, resulting in the establishment of a FIA-Network Relationship Map (NRM) model. The outcomes of this analysis demonstrate the importance and interaction degree for each key factor. Finally, based on the results of the FIA and NRM, suggestions were proposed for the development of the medical device development process ( Figure 1 ).

Research framework flowchart.
3.1. Participants
The questionnaire design was based on a literature review and content analysis of expert interviews. The questionnaire investigates the background of the respondents, including their age, work experience, professional knowledge, and the organization they work for, to confirm that the respondents conform to the current study on the medical device development process. After the questionnaire was distributed, a total of 65 valid samples were collected. They work in different professions related to health care and medical equipment. Among the medical device developers that we surveyed, 23 (36.0%) were from medical profession background, 32 (49%) were engineering expertises (including technical staff and managers), and 10 (15%) were from regulatory expertises. The respondents included 48 males (74%) and 17 females (26%). The average of seniority distribution of the respondents in the biomedical industry was 9.1 years, 78% are above 5 years, and 40% are above 10 years. The study also examined the level of medical device risk involved in the development experience of the respondents, including 5 (8%) who were involved in Class I risk, 33 (50%) who were involved in Class II risk, and 27 (42%) who were involved in Class III risk.
3.2. Content Analysis
The content analysis uses qualitative or quantitative data and involves methods of induction or deduction. Content analysis, which is also known as text or literature analysis, converts qualitative data into quantitative data for analysis. The value of content analysis lies in its utilization of system objective and quantitative methods to classify statistics. The hidden content of records can be systematically organized and visualized based on the narrative interpretation of the numbers in the categories. Content analysis methods are applicable when sorted verbal information is critical to the research [ 22 , 23 ].
Content analysis is an objective and systematic method for investigating and analyzing the content of documents and clearly describing the content of the communication. Moreover, it can analyze various languages and features in communication content [ 24 ]. The possibility of exploring a particular property of information can assist the prompt deduction of meaning. Additionally, the content analysis examines and analyzes communications to measure variables quantitatively, objectively, and systematically. Beneficial and simple, content analysis has been used in numerous aspects of scientific research for over six decades. The hypothesis of the present study states that the most frequently mentioned words reflect the biggest problem. Content analysis involves three steps, namely, unit coding, sampling, and validity analysis [ 25 , 26 ].
The experts interviewed in this study were experienced in the development, management, and use of medical devices and provided valuable advice during the interviews. Content analysis was used to analyze the interviews.
3.3. DEMATEL Method
The DEMATEL method can be used to study and solve complex and interwoven problem sets [ 27 ]. The DEMATEL method enabled the researchers to understand specific problems and interweave clusters of problems as well as to better identify possible solutions through hierarchical structures. Recent studies have used DEMATEL techniques to solve complex problems, such as the analysis of smart product service systems [ 28 ], probabilistic safety analysis of process systems [ 29 ], pharmaceutical manufacturing [ 30 ], and hospital performance management [ 31 ]. This method differs from traditional methods in that an NRM can identify interdependence among system elements through causal graphs. In this research, the DEMATEL method was applied to constitute NRMs to investigate whether the development processes of medical device design interact with one another or they are independent. The concept of the DEMATEL method is as follows [ 27 ]. Calculate the average matrix: first, organize actors through the questionnaire and obtain interactions among the factors. Each respondent will be asked to assess the direct impact of any two factors with an integer score ranging from 0 to 4, (0 = “no influence,” 1 = “low influence,” 2 = “medium influence,” 3 = “high influence,” and 4 = “extreme strong influence”). The next step is to establish the initial influence matrix: The impact between two pairs of factors will be compared in the survey questionnaire. X ij indicates the extent to which a respondent considered factor i affecting factor j , and the diagonal of the matrix shows the influence of the factor on itself, it will be set to 0 when there is no influence.
The next step is to establish the normalized direct-influence matrix: A normalized datum is the maximum of row vectors and the sum of column vectors. The normalization influence matrix is denoted by M , and the normalized datum is set to s . M and s can be calculated as follows:
To calculate the indirect-influence matrix, the indirect-influence matrix is set to IM. The indirect-influence matrix can be gained by directly affecting the value of matrix ( M ) calculated by the following equation:
To calculate the total-influence matrix, the value of the total-influence matrix can be obtained from the value of the direct-influence matrix, and the value of the indirect-influence matrix can be calculated using the following equations:
Subsequently, the structural relationship between the factors is analyzed.
The sum vector of the row value is d i , and the sum vector of the column value is r i . Then, if we let i = j , the sum vector of the row value plus the column value will be ( d i + r i ), which represents the center degree. If the sum of the row value plus the column value ( d i + r i ) is high; thus, the relationship among dimensions or criteria will be powerful. The sum of the row value minus the column value is ( d i − r i ), which indicates the extent of the reason. If d i − r i > 0, then the degree of influence on others is stronger than the degree of being influenced; otherwise, d i − r i < 0. Finally, the center degree ( d i + r i ) is taken as the X axis and the reason degree ( d i − r i ) is taken as the Y axis.
In this study, the structure influence relation diagram was drawn. Next, the relation diagram was divided into four quadrants by the average of the center and reason degrees. The distributions of the indices were observed on the influence network diagram, and the causality and core degree of the index were analyzed.
3.4. FIA Model
Martilla et al. originally proposed the importance-performances (IPA) model to verify the importance and performance of factors being investigated, thereby dividing the two axes into four quadrants and indices [ 32 ]. Based on this model, decision-makers can sort through and improve the relevant attributes of their products or services. The IPA model does not waste resources on inappropriate and informal strategies and has long been considered a simple and effective technique. The present study extended its analysis of the FI and II. As shown in Figure 2 , four frequency quadrants were constructed with frequency indicators based on the weighted survey provided by the respondents, and the impact indicates decision-makers to make strategic decisions. This study proposed four service improvement strategies for analyzing the four frequency and impact indicators.
- Priority: The first quadrant illustrates a high level of frequency and impact (H, H). This quadrant demonstrates that a factor has a high frequency and a high impact. Thus, medical device companies can prioritize solving this factor to strengthen their design development. In this research, we named this quadrant “Priority.”
- Investing resource: The second quadrant illustrates a low level of frequency and a high level of impact (L, H). The quadrant demonstrates that a factor has a high impact but does not reflect frequency. Therefore, medical device companies should invest resources in response to this factor. In this research, we named this quadrant “Investing resource.”
- Standstill: The third quadrant illustrates a low level of frequency and a low level of impact (L, L). This quadrant shows that factors situated in it have a low frequency and a low impact; thus, medical device companies can maintain their current status. In this research, we named this quadrant “Standstill.”
- Suspension: The fourth quadrant illustrates a high level of frequency and a low level of impact (H, L). This quadrant shows that the impact is not large, but the frequency is high. Medical device companies can suspend processing first. In this research, we named this quadrant “Suspension.”

3.5. NRM Analysis
The purpose of the DEMATEL method is to form a network diagram (i.e., an NRM). In addition, the method is mainly used to determine if factors interact or are independent, and the NRM is the final step in the DEMATEL method. The relationship between the degree and level of interaction of factors can be described by an easy-to-understand structure and a precise simplification of interdependence [ 33 ]. The NRM differs from the FIA model in the sense that it assigns and ranks factors based on specific characteristics. The NRM reveals the interrelationships among factors and evidence that provides additional important factors. A structural matrix and causal map can be used to show causality and impact, and the factors in a complex system can promote decision-making [ 34 , 35 ].
4. Data Analysis and Results
4.1. content analysis.
Based on the literature review, case studies and interviews with experts, the process of medical device development is divided into user action (UA), technical capability (TC), use environment (UE), and organizational characteristics (OC). To answer our research question (what are the challenges in the development of medical devices?), we interviewed experts with experience in medical device development in Taiwan. These experts come from a variety of medical device-related organizational backgrounds, including audit organizations, consulting firms, government agencies, and medical device companies. For this study, a minimum of six years of experience in the development or evaluation of multiple medical devices is required for someone to be called an expert. The respondents were interviewed face-to-face with informed consent in order to understand the key factors in the development of current medical devices. An overview of the stakeholders is provided in Table 1 . Three experienced coders are responsible for the verbatim coding of the interviews for content analysis. The UA, TC, UE, and OC profiles and 16 key factors were extracted from the interviews and cited in the literature or the expert interviews. In addition, these key factors were clearly defined and analyzed for reliability ( Table 2 ). Three coders performed the coding. Coders have experience in medical device innovation and underwent several rounds of practice coding with subsamples. Coders calculate the number of factors that each coder overlapped and then calculate mutual agreement and reliability. Disagreements were resolved after discussions and reassessments of the case to eventually arrive at a consensus. Table 3 shows that the average mutual agreement between the coders is 0.844, which is high. In addition, the reliability test presents that the reliability of the three coders interviewed is 0.942, and the values greater than 0.8 represent the high reliability of content analysis [ 36 ].
The interviewee's background information.
Assessment structure of medical device development.
Note: A: [ 14 ], B: [ 15 ], C: [ 16 ], D: [ 37 ], E: [ 38 ], F: [ 39 ], G: [ 8 ], H: [ 40 ], I: [ 41 ], J: [ 42 ], K: [ 17 ], L: [ 18 ], M: [ 43 ], N: [ 44 ], O: [ 19 ], P: [ 20 ], Q: [ 45 ], R: [ 46 ], S: [ 47 ], T: [ 21 ], U: [ 9 ].
Reliability and mutual agreement between coders.
0.844 indicates high agreement between coders for content analysis. 0.942 is greater than 0.8 indicating high reliability of content analysis.
4.2. FIA-NRM Model
The purpose of FIA is to conduct placement positioning based on the dimensions and the factors FI and II such that the medical device developers can have an adequate command of the frequency and impact of each dimension or factor. To build the FIA models, the average values of the weights (from 0 to 10 points) provided by the respondents to the criteria are calculated and standardized using standard deviation. Each criterion has one frequency value and one impact value, which helps determine its position in the FIA model. NRMs are developed using the DEMETAL method to present the causal relationship and the degree of impact between the dimensions and barriers in a complex system, which can facilitate the decision-making process. Criteria with high ( d + r ) values have strong relationships with other criteria, whereas those with low ( d + r ) values have weak relationships with other criteria. Furthermore, criteria with a positive ( d − r ) can influence other criteria, whereas those with a negative ( d − r ) have a high chance of being influenced by others.
Based on the reliability and validity analysis, the Cronbach's alpha of the primary dimension is 0.959, the Cronbach's alpha of UA is 0.886, the Cronbach's alpha of TC is 0.872, the Cronbach's alpha of UE is 0.906, and the Cronbach's alpha of OC is 0.871. The results show that the research questionnaire demonstrates high reliability ( Table 4 ).
Reliability and validity analysis.
Note. Cronbach's α values show that α < 0.35 is lowly creditable, 0.35 < α < 0.7 is moderately creditable, and α > 0.7 is highly creditable.
4.2.1. Primary Dimensions
Primary dimensions, including UA, TC, UE, and OC, were analyzed in the FIA model, which is characterized by levels of impact and frequency. Figure 3 and Table 5 reveal that TC has a high impact and high frequency; thus, priority should be given immediately. OC and UA have a high impact but low frequency; thus, considerable resources and efforts should be invested to abolish factors. Finally, UE has a low impact and low frequency; thus, developers can maintain standstill action. Therefore, developers should prioritize the following order of the dimensions: TC ⟶ OC ⟶ UA ⟶ UE. As for the NRM model, Figure 4 and Table 5 indicate that UA demonstrates the highest ( d + r ) value and the strongest connection with the other dimensions. Furthermore, UA has a positive ( d − r ) value, and thus has a remarkable impact on the other dimensions. For further observations on the causal relationships between the primary dimensions, Table 6 provides data on their net influence. Table 6 points out that OC influences all the other dimensions, TC influences UA and UE, and UE influences UA.

Main dimensions' FIA model of medical device development.

Main dimensions' NRM model of medical device development.
Statistical analysis and strategy for main dimensions.
Note. L stands for “low” and H stands for “high.”
Net influence matrix for primary dimensions.
Figure 4 shows that four improvement pathways exist via NRM analysis, that is, OC ⟶ UA, OC ⟶ TC ⟶ UA, OC ⟶ UE ⟶ UA, and OC ⟶ TC ⟶ UE ⟶ UA. Tables Tables5 5 and and6 6 demonstrate that the ranking of the FI is TC > UE > OC > UA, and the ranking of the II is TC > UA = OC > UE. To find a possible pathway, a dimension with a high rank is used to affect a dimension with a low rank. For example, in FI, the second pathway, that is, TC (ranked 1) can improve UA (ranked 4), and this pathway will be accepted. The remaining pathways also follow this logic. Four solvable pathways exist in the FI, and four solvable paths likewise exist in the II. Next, we find four overlapping solvable pathways, as shown in Table 7 . Table 7 summarizes the improvement paths and recommended pathways that medical device developers can follow to solve the main dimensions of medical device development.
- Developers should efficiently take investing resources to improve OC to determine UA.
- Developers should efficiently take investing resources to improve OC, then take priority action to ameliorate TC to determine UA.
- Developers should efficiently take investing resources to improve OC, then take standstill action to define UE to determine UA.
- Developers should efficiently take investing resources to improve OC, take priority action to ameliorate TC, then take standstill action to define UE to determine UA.
Recommended pathways for solving main dimensions.
4.2.2. User Action Dimensions
Four categories comprised of UA, namely, the user needs considerations (UA1), training course (UA2), empirical cognitive ability (UA3), and physical and mental health (UA4). Table 8 and the FIA model in Figure 5 indicate that UA1 and UA2 have a high frequency and high impact. Hence, developers should prioritize solving these two categories immediately. As UA3 has a high impact but low frequency, developers can assess the investment of resources. Finally, the low levels of impact and frequency of UA4 suggest standstill action. Developers are recommended to prioritize the following order of the factors: UA1 ⟶ UA2 ⟶ UA3 ⟶ UA4.

FIA model for UA.
Statistical analysis and strategy for UA.
As for the NRM model, Figure 6 reveals that UA4 influences all the other UA categories, UA1 influences UA3 and UA2, and UA3 influences UA2.

NRM model for UA.
Figure 6 and Table 8 show that the ranking of the FI is UA1 > UA2 > UA3 > UA4, and the ranking of the II is UA1 > UA3 > UA2 > UA4. Two solvable pathways are observed in the FI, and three solvable paths are seen in the II. Next, we find two overlapping recommended pathways: UA4 ⟶ UA1 ⟶ UA2 and UA4 ⟶ UA1 ⟶ UA3 ⟶ UA2.
4.2.3. Technical Capability Dimensions
There were four categories that comprised TC, namely, user interface design (TC1), competitive products (TC2), calibratable maintenance (TC3), and label warning (TC4). Table 9 and the FIA model in Figure 7 indicate that TC1 and TC4 have a high frequency and high impact. Hence, developers should prioritize solving these two categories immediately. As TC2 has a high impact but low frequency, developers can assess the investment of resources. Finally, the low impact and high frequency of TC3 suggest the suspension of action. Developers are recommended to prioritize the following order of the factors: TC1 ⟶ TC4 ⟶ TC2 ⟶ TC3. As for the NRM model, Figure 8 reveals that TC1 influences all the other TC categories, TC2 influences TC3 and TC4, and TC3 influences TC4.

FIA model for TC.

NRM model for TC.
Statistical analysis and strategy for TC.
Figure 8 and Table 9 demonstrate that the ranking of the FI is TC4 > TC3 > TC1 > TC2 and the ranking of the II is TC1 > TC2 > TC4 > TC3. Two solvable pathways exist in the FI and four solvable paths exist in the II. We find two overlapping recommended pathways: TC1 ⟶ TC2 ⟶ TC4 and TC1 ⟶ TC2 ⟶ TC3 ⟶ TC4.
4.2.4. Use Environment Dimensions
UE is comprised of four categories, namely, intended location (UE1), public safety protection (UE2), hygiene requirements (UE3), and device usage time (UE4). Table 10 and the FIA model in Figure 9 indicate that UE1 and UE4 have a low impact but high frequency, thereby suggesting the suspension of action. The low levels of impact and frequency of UE2 and UE4 suggest that standstill action should be taken. Developers are recommended to prioritize the following order of the factors: UE1 ⟶ UE4 ⟶ UE2 ⟶ UE3. As for the NRM model, Figure 10 reveals that UE1 influences all the other usage barriers, UE4 influences UE2 and UE3, and UE2 influences UE3.

FIA model for UE.

NRM model for UE.
Statistical analysis and strategy for UE.
Figure 10 and Table 10 demonstrate that the ranking of the FI is UE4 > UE1 > UE2 > UE3, and the ranking of the II is UE3 > UE1 > UE2 > UE4. Three solvable pathways are seen in the FI, and three solvable paths exist in the II. We found three overlapping recommended pathways: UE1 ⟶ UE4 ⟶ UE3, UE1 ⟶ UE2 ⟶ UE3, and UE1 ⟶ UE4 ⟶ UE2 ⟶ UE3.
4.2.5. Organizational Characteristic Dimensions
OC had four categories, namely, management culture (OC1), team communication (OC2), resource allocation (OC3), and regulatory standards (OC4). Table 11 and the FIA model in Figure 11 indicate that OC4 and OC2 have a high frequency and high impact. Hence, developers should prioritize solving these two categories immediately. The low levels of impact and frequency of OC1 and OC3 suggest that standstill action should be taken. Developers are recommended to prioritize the following order of the factors: OC4 ⟶ OC2 ⟶ OC3 ⟶ OC1. As for the NRM model, Figure 12 provides data on their net influence. Table 11 presents that OC1 influences all the other OC1 categories, OC2 influences OC4 and OC3, and OC4 influences OC3.

FIA model for OC.

NRM model for OC.
Statistical analysis and strategy for OC.
Figure 12 and Table 11 demonstrates that the ranking of the FI is OC4 > OC2 > OC3 > OC1, and the ranking of the II is OC4 > OC2 > OC3 > OC1. Three solvable pathways exist in FI, and three solvable paths are observed in II. We identified three overlapping recommended pathways: OC1 ⟶ OC2 ⟶ OC3, OC1 ⟶ OC4 ⟶ OC3, and OC1 ⟶ OC2 ⟶ OC4 ⟶ OC3.
Recommended pathways based on the results of each of the above factors and tables and planning for the overall improvement path are shown in Table 12 . The recommended improvement pathway order is OC ⟶ TC ⟶ UE ⟶ UA. Medical device developers should examine and analyze each factor. A range of informal and formal organizational processes can influence user considerations, user interfaces, and UE in the development of medical devices. Moreover, adopting a formal decision-making process can help medical device developers develop an integrated and reflective approach to improve business decisions and quality end products.
Summary of improvement pathways.
5. Discussion
This study clarifies the key factors of the medical device development process from the user and stakeholder perspectives. In addition, this study analyzes the frequency and impact of these critical factors on the medical device development process and suggests strategies for improvement.
5.1. Theoretical Implications
This study fills the research gap in key factors that affect the development of medical devices and improvement paths. The FIA results indicate that medical device development stakeholders consider OC as the focus of medical equipment development. Consistent with the views of medical device regulatory authorities, the results demonstrate the importance of the safety and effectiveness of medical devices, including well-designed human-computer interface interaction based on user needs and conditions, and clearly defined product use information that includes mentioning warnings (based on product functions). Medical devices must comply with regulations before they can be marketed, and a recall mechanism should be in place in case of efficacy and safety concerns after marketing. Inappropriate medical devices will be deregulated or banned from the market because they often cause harm to end-users [ 41 ].
Although medical device development stakeholders consider UA and OC as infrequent problems, the two dimensions nonetheless exerts a large impact on medical devices. Numerous studies have noted that shortcomings still exist in the design of medical devices in terms of usability from users' perspectives, such as balancing conflicting user needs and ethical privacy [ 4 , 48 ]. Medical device developers should identify priority input considerations as early as possible to satisfy user needs and provide education, training, and safety guidelines from the users' perspective [ 49 ]. This recommendation is also consistent with stakeholders' views that UA considerations should focus on satisfying user needs and improving training courses, whereas the assessment of user background and individual physical and mental status is difficult and not a priority in the product development process. OC aspects, including regulatory standards and team communication, from medical device design to market entry, are also important in the development and profit of medical devices. Team communication within organization is critical [ 50 ]. Moreover, owing to the particularity of medical devices, regulatory standards have become a key consideration in the marketing of medical devices. Medical device enterprises should familiarize themselves with national regulations as well as the economic status and social backgrounds of their targeted market as early as possible [ 9 ] and develop their product listing process and market plans. Finally, medical device company stakeholders believe that UE is not a priority in the product development process. This finding may be due to the strict regulatory mechanisms of medical devices for product safety specifications; thus, parameter settings and range have been applied to most environmental factors.
5.2. Practical Implications
Medical devices make resource input in its development process far higher than that of general products. Compared with large enterprises, SMEs are disadvantaged in terms of risk control, manufacturing, and operation performance owing to insufficient resources [ 51 ]. This finding has made it necessary for numerous SMEs that manufacture medical devices to evaluate resource planning strategies and develop appropriate paths for product development and healthcare benefits in the context of limited resources [ 52 , 53 ]. Based on the views of stakeholders on the development of medical devices, this study proposes development order and path suggestions in the development of medical devices ( Figure 13 ). The results of this study suggest that medical device development strategies should improve management culture and team communication within the organization and allocate development resources after confirming medical device regulatory standards. After confirming the feasibility of development, medical device developers need to consider the user interface design in terms of technical capability, establish calibration and maintenance standards, and label warnings on their products. Next, medical device developers need to consider the impact of the surrounding environment on the device, including the location, time of use, protective facilities, and hygiene requirements. Finally, even though medical regulations have established the safety and usability of medical devices, medical device developers must still take into consideration the unique circumstances of possible users.

Medical device development strategy.
The development of medical devices is usually for start-up teams or SMEs. The establishment of a climate of intense collaboration and communication between different areas of the organization not only facilitates motivated new projects and rapid decision-making, but also focuses on user needs from concept to disposal of the product lifecycle and integrates the development process, which is an important basis for the development of medical devices [ 54 ]. Due to the complexity of the multisite, multiperson, and multidevice context of many medical interactions, the ensuing user behaviour can have a range of implications for the effectiveness of medical procedures. Developers need to design user interfaces based on technical features, in particular to understand the ergonomic impact of products and clinician/nursing staff interactions with patients based on information from competing products, and to establish product maintenance and warning standards [ 55 ]. The clinical environment usually involves at least two participants in the interaction (clinician and patient) and there are often many complex environmental factors that affect the overall procedure or task, such as the conditions of use of the device (e.g., portability, manoeuvrability, conflict of existing equipment and use of power outlets), the physical environment (e.g., the impact of bedrail design on patient behaviour), and the size of the space available may all limit the usability of the medical device. Developers should therefore also consider the impact of environmental factors on the use of the device when assessing the overall outcome of the device design. Finally, although all of the above factors are met, a medical device is considered marketable. However, the findings of this study suggest that it would be helpful if the development team could take into account the user's condition, including physical and mental health, needs, and training. For example, the packaging of disposable devices may affect the time and efficiency pressures on medical staff, while the sensitive clinical nature of ultrasound is crucial to the physical and psychological comfort of patients. In addition, in the case of long-term health outcomes, other factors of the patient (age, clinical condition, and medication effects) may be no less influential than the design of the device [ 55 ]. This inside-out process of influence is a key in the development of numerous enterprises [ 5 , 56 ] and covers various fields. Thus, in the present study, the proposed development paths of medical devices from the perspective of stakeholders can be seen as logical and valuable.
6. Conclusion
This study uses content analysis and FIA-NRM to discuss stakeholders' views on key factors in medical device development. This study summarizes important factors in the development of medical devices and the views of various stakeholders. This research suggests that the development of medical equipment should start with OC and strengthen TC. Next, according to the evaluation indicators of this study, medical device developers can consider UA and UE strategies and improve functional design, product safety, and clinical application planning with optimal resource allocation. In the future, this study will be able to incorporate input from other stakeholders, including healthcare providers, venture capitalists, and government agencies. It will also be able to conduct case studies on different medical device categories. In the long run, the medical device development strategies developed in this study can benefit the medical industry, health care policy, and national development.
Acknowledgments
The authors would like to thank the laboratory members Mr. Wei-Zhi Lu, Miss. Hui-Chi Wei, and Hsin-Hui Chiu for their help in coding and questionnaire collection. The authors would like to thank Dr. Shun-Min Wang for his expertise in contributing to the construction of the evaluation framework and the research respondents recruited, which helped to relate our research to real-world practice. This research was supported by the Ministry of Technology and Science under grant numbers 108-2221-E-006-063 and 109-2410-H-006-045-MY2 and the Medical Device Innovation Center (MDIC).
Data Availability
Conflicts of interest.
The authors declare that they have no conflicts of interest.
- MDR CER Writing
Systematic Literature Review
- Literature Review Templates
- Full Service PMS
- Vigilance Monitoring and Reporting
- PMCF Surveys Solution
- Clinical Evaluation Doc Reviews
- IVDR Writing
- FDA Submissions
- Consulting Services
- SLR Software – CiteMed.io
- EU MDR Templates for CER and PMS
- Free Downloads
- Featured In …
- Testimonials
Your On-Demand Clinical Evaluation Team
Full Clinical Evaluation Report (CER) services and the Industry’s Fastest Literature Review for EU MDR.
Ace your audits, maximize your team’s time and budget.
Meeting Deadlines
Guaranteed approval, vip service, peace of mind, problems we solve, clinical evalution, get support from seasoned cer writers., done for you by experts, or use our industry leading software., post market surveillance and vigilance, handle your pms and vigilance needs year after year with ease., emergency damage control, need to organize audit responses fast we’ve got you., regulatory templates, small team and tight budget use our constantly updated template packages., training courses, learn from qa/ra practitioners who have been there before., regulatory news updates newslatter, stay on-top of the latest industry news. free., how we work, done for you.
Don’t have the time, or a particular type of submission experience?
Our regulatory team is stacked with experience, and if you look closely the occasional battle scar.
Done With You
Looking for on-demand help when you need it? (and only when you need it)
Sometimes an expert review of a doc or SOP is all it takes to polish a submission. Other times you might need more.
Regardless of your level of assistance required, our team is here to help when you need it and get out of the way when you don’t.
Do It Yourself
Under budget, or a freelance writer looking to get to next level? Our training programs and templates will get you there.
Are You a Fit?
-who we serve, medical device manafacturers.
Small/medium
Consultants
Medical Writers
Our Previous Clients
Our track record speaks for itself, but we won’t say no to a little extra flattery from clients!
CEO of Device Manufacturer CERs are the bane of my existence! Being able to pass them all off to CiteMed’s hands was a massive sigh of relief for me and my employees.
Quality consultant eu mdr project lead the writers really got into our device, and listened to feedback. the level of detail was a huge help in our cer drafting process., vp of quality device manufacturer i loved the weekly update reports with just the right amount of detail. the ‘timeline risk rating’ was really helpful and made us feel comfortable throughout the project, director of qa/ra device manufacturer our lit review project was completed on-time, in only 3 weeks despite some bumps, citemed delivered, regulatory engineer device manufacturer it was a huge advantage being able to leverage their experience on our final round of audit responses. thanks again, vp of regulatory affairs their approach to slr is sound, and consistent. i love knowing that updates will be handled for us the next few years, jen s. quality director i was blown away by their responsiveness and service. they understood my urgency and got on the phone the same day i inquired., the citemed edge.
What makes our approach unique is our blend of world class regulatory talent, leading regulatory software tools, and (almost) obsessive customer service. Here are some of the highlights:
High quality Systematic Literature Review on Industry Leading Timelines
CER Team Boasting 100% Acceptance Rate in Europe
Systematic Literature Review Software saving teams over 20+ hours per Literature Review
Dedicated Account Reps to manage your project, and be your single point of contact.
Shortest time to Proposal, and Project Kick Off. Zero time wasted, all to improve your delivery timeline.
Your Literature-Driven Regulatory Process
Consistent and effective Literature Search/Review strengthens your entire regulatory workflow.
Cite Medical Literature Search Dashboard
Our team works with our proprietary platform to search, process, and store all scientific literature related to your devices.
Clinical Literature
We search globally and utilize an extensive list of databases such as PubMed Europa, Embase, Cochrane, Clinicaltrials.gov, and more.
Vigilance and Adverse Events/Recall
We search and record data from every country that maintains a public database for Adverse Events and Recalls.
Cite Medical Literature Review
The Literature Review document you receive is comprehensive and designed for easy integration into your CER, PMCF, or PSUR.
Clinical Evaluation Report
Whether it’s a first time submission or an update, bolster your CER with a bullet-proof Systematic Literature Review.
PMCF Report
While rarely the only requirement, an update demonstrating strong support from Clinical Literature is a great addition to your PMCF commitments.
Periodic Safety Update Report
Our Literature Reviews bolster your PSUR narrative and showcase your team’s diligence in updating your safety information and benefit-risk statement.
Our Trending Publications
Navigating global medical device regulations.
The CiteMed Team 2024-02-22T01:57:33+00:00 February 20th, 2024 |
Understanding the Landscape of Global Medical Device Regulations The global landscape of medical device regulations is as diverse as it is complex. Navigating through these regulations requires a deep [...]
Navigating Medical Device Safety, Regulation, and Adverse Europe
The CiteMed Team 2024-02-14T00:40:47+00:00 February 13th, 2024 |
Adverse Events in Europe The landscape of medical device safety in Europe is significantly influenced by the occurrence and management of adverse events. An adverse event in this context [...]
Medical Device Regulations – Links You Should Be Aware Of
The CiteMed Team 2024-01-29T18:43:44+00:00 January 29th, 2024 |
Medical device regulations are no joke. It's pretty important to get the regulations right, since even a single missed data, if significant enough, can result in years of delay [...]
Before You Hire a Clinical Evaluation Consultant: Read This
The CiteMed Team 2024-01-23T09:47:03+00:00 January 23rd, 2024 |
Thinking of Hiring a Clinical Evaluation Consultant? So it's finally time to either start that new Clinical Evaluation Report (CER) project for Europe, or remediate your current documents based [...]

Medical Device Regulation Clinical Evaluation Literature Review – Part Three

Appraising and evaluating the search results
In our previous blog, we focused on performing a successful database search and doing the first screening of search results through titles and abstracts.
In this final, third part of our three-part series, we would like to show you how to do the second level of screening, i.e., full-text analysis, and capture relevant data for the clinical evaluation report.
If you are interested to join our newsletter email list and receiving our template for the Clinical Evaluation Report (CER), please write us directly .
Step One: Find and gather the full text of selected documents
In our previous post , we talked about the first level of screening the results. On that stage we excluded publications that are obviously not relevant on the basis of the contents of the abstract or title. We left the ones that are, at first glance, important for our Literature Review.
In this step, your aim will be to find the full texts of all the references you’ve selected.
According to MEDDEV 2.7/1 Rev. 4 and MDCG 2020 13, the full-text papers and documents should be obtained for the appraisal stage.
Step Two: Evaluating the Relevance
Read and critically appraise the full text of each article you selected. It is crucial to determine if the identified data is relevant for the medical device under evaluation. According to MEDDEV 2.7/1 revision 4, when evaluating the relevance of the full-text articles, it is essential to consider whether the data are intended to:
- directly demonstrate adequate clinical performance and clinical safety of the device (pivotal data), or
- serve as an indirect supportive role.
The pivotal data must have the quality to demonstrate the clinical performance and the safety of the device under evaluation. Therefore, it must be generated with the device in case or with an equivalent device used for its intended purpose.
For more information on this topic, see the MEDDEV 2.7/1 Appendix A6, Appraisal of clinical data – examples of studies that lack scientific validity for demonstration of adequate clinical performance and/or clinical safety and MEDDEV 2.7/1 Appendix A1, Demonstration of equivalence.
Data that are not pivotal are generally appraised for their contribution for identifying and defining:
– the current knowledge/ state of the art in the corresponding medical field, to define acceptability criteria for the evaluation of the benefit/risk profile and of specific side-effects of the device under evaluation;
– hazards; individual case reports may be used for identification of new and previously unknown hazards that are associated with the device;
– the validity of criteria used for the demonstration of equivalence (if equivalence is claimed) etc.
Questions that help determine whether data are relevant are listed in Section 9.3.2.c of MEDDEV 2.7.1 rev. 4, we provide here in the summarized form:
- Are the data relevant to the intended purpose or claims of the device?
- Suppose the data are relevant to specific aspects of the intended purpose or claims. Are they applicable to particular device model, user group, medical indication, age, gender, the severity of the condition, or range of time?
- To what extent are the generated data representative of the device under evaluation?
- What aspects are covered?
If the article is relevant
If you decide that an article is relevant, the next step is to weigh its clinical contribution. Unfortunately, there is no single method for weighting clinical data. You should choose a rating system you believe it is the most adequate for the device under evaluation (for example, OCEBM, SORT, GRADE, or other rating scales.)
The scientific validity of the clinical data set: Level of Evidence of the publication
If the article is not relevant
You are expected to provide a rationale for each article excluded from the analysis. The reasons might vary:
- Different devices, Different indications, Different population
- Inappropriate number of patients, duration, no statistical analysis,
- The non-comparative study, inappropriate clinical outcome
- The preclinical study not on the device under evaluation
- Socio-economic assessment, a cost-effectiveness study
- Comments on an article, poster, abstract, video not on the device under evaluation
- Non-peer-reviewed opinion/journal not on the device under evaluation
- Foreign language not generally understood
- Out of the scope
- Single Case study report without any relevant safety information
- The article is duplicated (same author, same study…)
You can add the table with the codes for the exclusion rationale:
Important! The sum of included and excluded articles should sum up to the number of articles you’ve got by the literature search.
If the available data is limited
According to MEDDEV 2.7/1 revision 4, in exceptional situations, when an evaluation is based on limited data, you can describe and justify that in the clinical evaluation report. Please, see additional information and specific considerations in Appendix A8 (Devices for unmet medical needs – aspects to consider).
Step Three: Write the Literature Search Review Report
Finally, you can write the Literature Search Review Report which contains the results of your search that address safety, performance, benefits to the patient, side-effects, adverse reactions, state of the art of the device in question.
A CER literature review report should include Introduction (Purpose), Objectives, Method, Search results, Analysis, and Conclusion.
For Introduction, Objectives and Methods you should use the info pre-defined in your literature review protocol. Provide a summary of the searches that were performed, the number of results, and the number of articles excluded. The analysis of the reviewed literature should contain a comprehensive summary and analysis of the information identified during the literature review process, both favorable and unfavorable, and it should address each of the CER literature review objectives. In Conclusion, you should provide a summary to indicate how the objectives of the literature review have been met.
At the end, you should add Appendices to your Report: Bibliography, Detailed data analysis tables and spreadsheets, Literature search results list and detailed criteria for the article exclusions.
BioReg Services specializes in systematic literature searches and reviews in compliance with the MDR Regulation (EU) 2017/745. Our team can design a comprehensive literature search protocol with complete documentation of the literature screening and data extraction process. We can obtain clinical evidence for the literature review for your new medical device, or we can update the literature review of a CE-marked or legacy device to create an MDR compliant document. Don’t hesitate to contact me directly: [email protected].
BioRegards,

Previous post
Medical Device Regulation Clinical Evaluation Literature Review - Part Two
31 tips for successful mdr clinical evaluation & cer, you may also like.

Israel Device Declaration Registration

Israel Medical Devices Registration

ISO 13485 for Device Manufacturers
IMAGES
VIDEO
COMMENTS
The MDR regulation outlines specific requirements for the literature review — a robust process of executing evidence-based research queries that describe medical device product safety, benefit, and risk. Literature reviews are comprehensive searches of published information found in scientific studies related to medical devices on the market.
Appraisal of the clinical data. 4. Analysis and conclusions generated from the clinical data. 5. Informatic tools. 6. Process flow. Conclusions. Catarina Carrão, freelance medical writer on Kolabtree, outlines the importance of literature reviews for medical devices and best practices to follow.
Medical devices (MDs) as defined by the World Health Organization (WHO) (WHO 2012) are equivalent to the medical product terminology used by the Brazilian Health Regulatory Agency (ANVISA) (ANVISA 2010) and to the medical equipment definition stated by the Brazilian Health Ministry (MS) (ANVISA 2013a).In this review, we use the WHO terminology and refer to MDs hereafter.
We conducted a comprehensive literature search on January 2, 2020. The following electronic databases were searched with the assistance of an information specialist at the medical library: PubMed, EMBASE, Scopus, and the Cochrane Library. The review was limited to texts published in English between 2015 and 2019 for which abstracts were available.
In addition, a narrative literature review was conducted for a synthesis of the characteristic differences and challenges of HTA in Medical Devices. The literature search was performed using PubMed, Embase, and Web of Science. We included relevant empirical studies or reviews discussing the use of HTA for medical devices.
The systematic literature review was performed by applying the published standard, ... Firstly, the general terms of medical equipment or medical device or other specific equipment categorised under these general terms were mentioned in the title and keywords. Secondly, an indication of the quantitative method in assessing the medical equipment ...
Expert Review of Medical Devices [ISSN 1743-4440]; [e-ISSN 1745-2422] is a MEDLINE-indexed, international peer-reviewed journal providing commentary, analysis and debate for all professionals involved in research, development, testing and clinical use of devices.. Expert Review of Medical Devices aims to address the needs of the device research community by providing a comprehensive body of ...
Highlights What is already known. Systematic reviews of medical devices require systematic searches. Whilst the methods of searching are similar to other types of systematic review, for example reviews of medicines, the practice of designing a search and deciding where to search for medical devices present their own challenges.
Literature review We performed an extensive literature review for papers pub-lished before February 2016 for publications in English, Portuguese, and Spanish that covered the search query "us-ability" AND "medical device." The search was performed using a metasearch engine provided by the Brazilian
The decision to claim equivalence (or not) will be one of the factors that affects your literature review. The literature review is the means by which you obtain indirect clinical data. Many companies, especially if they are evaluating a low-risk device, will rely heavily on data from literature searches, as they won't be carrying out any ...
A comprehensive, objective, and thorough systematic literature review to describe the general State of the Art and identify all relevant clinical safety, performance, and usability data of the device under evaluation (and/or equivalent device) should comply with strict regulatory guidelines as per EU MDR 2017/745 and MEDDEV 2.7/1 Rev. 4.. In this blog post, we would like to share with you the ...
The Medical Device Coordination Group says the same. "For general guidance on a literature search, see MEDDEV 2.7/1 Revision 4, A5. Literature search and literature review protocol, key elements" ... In section D, the MDCG 2020-13 states that multiple databases should be used to minimize bias in the literature review.
It is argued that better selection of usability methodologies in MDDD should be based around three factors: application of current technical standards on usability, usage of health technology assessment literature, and consideration of ethics-related specificities of MD design. Purpose User involvement during medical device (MD) development and usability engineering techniques may help reduce ...
The European Medical Device Regulation (MDR) outlines the requirements for the literature review needed for the clinical evaluation of medical devices. You need a robust process for performing ...
Methods and Findings. We performed a systematic review to find empirical studies evaluating medical device regulation in the US or EU. We searched Medline using two nested categories that included medical devices and glossary terms attributable to the US Food and Drug Administration and the EU, following PRISMA guidelines for systematic reviews.
Automating the literature review process, though, brings three important benefits to medical device companies preparing CER and PER submissions. Do More Faster and Smarter. MDR and IVDR requirements force medical device manufacturers to increase the frequency, traceability, and overall documentation of clinical and product evaluation reports.
A literature search & review is a pivotal component of medical device Clinical Evaluation. According to MedDev 2.7/1 rev 4, they are essential for identifying safety and/or performance data relating to both the clinical background (State-of-the-Art) and to the subject device itself. The Clinical Evaluation Plan (CEP) should set out a carefully ...
UPDATE February 2023: The FDA added 3 new safety summary reports to the list of safety summaries below: The vast majority of patients implanted with medical devices have no adverse reactions. The ...
Step Three: Write a Literature Search and Review Protocol. A Literature Search and Review Protocol should contain: Research question. Databases that will be used. Terms that will be used. Inclusion/exclusion criteria. Search methods used in the literature evaluation. How the duplication of data from multiple sources will be addressed.
A clinical evaluation is required for all medical devices according to the MDR. The main task of a clinical evaluation is to identify pertinent data in relation to your software device and similar ones. This will help you to prove the intended use of your software device and the clinical claims, that your software device is safe, presents no risks or the benefits outweigh the risks, and ...
Creating an EU CER Literature Review Protocol and Reviewing Medical Device Clinical Data. ... (CER) is the culmination of a monumental effort to conduct literature searches, find/review literature, and/or conduct original clinical investigations. Data must be sourced, appraised, analyzed, and then summarized into your CER. This final process ...
Although many studies have examined the medical device development process, there has been no systematic and comprehensive assessment of the key factors affecting medical device development. This research synthesized the value of medical device industry stakeholders' experiences through a literature review and interviews with industry experts.
Cite Medical Solutions is the leader in EU MDR (medical device regulatory) consulting, high-quality literature search, and post market surveillance systems. ... Systematic Literature Review Software saving teams over 20+ hours per Literature Review. Dedicated Account Reps to manage your project, and be your single point of contact.
We left the ones that are, at first glance, important for our Literature Review. In this step, your aim will be to find the full texts of all the references you've selected. According to MEDDEV 2.7/1 Rev. 4 and MDCG 2020 13, the full-text papers and documents should be obtained for the appraisal stage. Step Two: Evaluating the Relevance.