A .gov website belongs to an official government organization in the United States.
A lock ( ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
- Outbreak Linked to Frozen Organic Strawberries
- Outbreak Linked to Fresh Organic Strawberries
- Prevention Approaches for Viral Hepatitis in Gay and Bisexual Men
- Global Viral Hepatitis
- Viral Hepatitis Outbreaks
- Viral Hepatitis
- Serology Training
- Hepatitis Posters Resources
- Viral Hepatitis Among Specific Populations
- Reporting HBV & HCV Infections in Repeat Blood Donors
- Health Care Outbreak Toolkit
- Planning, Progress, and Policy
- Funded Partners and Programs
- Influential Scientific Information
- Laboratory Testing
Related Topics:
- View All Home

Hepatitis A
Hepatitis b, hepatitis c.
- Viral Hepatitis Statistics & Surveillance
Clinical Overview of Viral Hepatitis
- Many people with a viral hepatitis infection do not have symptoms and are unaware of their infection.
- Chronic hepatitis B and chronic hepatitis C can cause serious health problems, including liver damage, cirrhosis, liver cancer, and even death.
- Hepatitis A and hepatitis B are vaccine-preventable, and hepatitis C can be cured with treatment.

Overview of Viral Hepatitis in the US
Each year, tens of thousands of people acquire a viral hepatitis infection in the United States. It is a serious public health threat that kills thousands of Americans annually and is a leading cause of liver cancer. Hepatitis A and hepatitis B are vaccine-preventable, hepatitis B can be treated, and hepatitis C can be cured.
The US has the opportunity and the responsibility to eliminate viral hepatitis as a public health threat. By working with clinicians and their patients, we can collectively achieve this goal. See below for further information on the cause, incidence, and prevalence of the most common types of viral hepatitis in the US.
- Hepatitis A is caused by the hepatitis A virus (HAV).
- There were an estimated 4,500 acute infections in 2022.
- Hepatitis B is caused by the hepatitis B virus (HBV).
- There were an estimated 13,888 acute infections in 2022.
- There were an estimated 640,000 adults with chronic HBV infection during January 2017–March 2020.
- Hepatitis C is caused by the hepatitis C virus (HCV).
- There were an estimated 67,400 acute infections in 2022.
- There were an estimated 2.4 million people - and as many as 4 million people - with HCV infection from 2017–2020. 1
Types and strains
Viral hepatitis is most commonly caused by three viruses: hepatitis A, hepatitis B, and hepatitis C.
Likelihood of symptomatic acute infection
- Less than 30% of children that are younger than 6 years old have symptoms, which do not typically include jaundice.
- More than 70% of older children and adults have jaundice.
- Most children younger than 5 years old do not have symptoms.
- People aged 5 years and older develop symptoms in 30%–50% of cases.
- Newly infected, immunosuppressed adults generally do not show symptoms.
- Jaundice might occur in 20%–30% of people.
- Nonspecific symptoms like loss of appetite, fatigue, or abdominal pain might be present in 10%–20% of people.
Potential for chronic infection after acute infection
Hepatitis A does not progress to chronic infection.
Chronic HBV infection develops in approximately:
- 90% of infants who acquired HBV infection perinatally or at birth.
- 30% of children who acquired HBV infection between 1–5 years old.
- 5% of people who acquired HBV infection as adults.
Chronic HCV infection develops in most people.
- Most people with acute disease recover with no lasting liver damage.
- Death is uncommon but occurs more often among older people and/or those with underlying liver disease.
- Acute illness is rarely fatal.
- 15%–25% of people with chronic infection develop chronic liver disease, including cirrhosis, liver failure, or liver cancer.
- Approximately 5%–25% of people with chronic hepatitis C will develop cirrhosis over 10–20 years.
- People with hepatitis C and cirrhosis have a 1%–4% annual risk for hepatocellular carcinoma.
Incubation period
Many people with viral hepatitis do not have symptoms and are unaware of their infection. If symptoms occur with an acute infection, they can appear anytime from 2 weeks to 6 months after exposure. Symptoms of chronic viral hepatitis can take decades to develop and are typically the same as those for acute infection.
Hepatitis A has an incubation period of 15–50 days, with an average incubation period of 28 days.
Hepatitis B has an incubation period of 60–150 days, with an average incubation period of 90 days.
Hepatitis C has an incubation period of 14–182 days, with an average incubation period of 14–84 days.
How it spreads
The different strains of viral hepatitis are transmitted through several possible exposures:
Hepatitis A is transmitted via fecal-oral route. This can happen through:
- Ingestion of contaminated food or water.
- Close person-to-person contact with a person who has HAV infection.
- Sexual contact with a person who has HAV infection.
Bloodborne transmission of HAV is uncommon.
Hepatitis B is transmitted via percutaneous, mucosal, or nonintact skin exposure to infectious blood or other body fluids. HBV is concentrated most highly in blood, and percutaneous exposure is an efficient mode of transmission.
HBV is transmitted primarily through:
- Childbirth.
- Sexual contact.
- Sharing contaminated needles, syringes, or other equipment used to prepare or inject drugs.
Less common transmission routes for HBV include:
- Needle-sticks or other sharp instrument injuries.
- Organ transplantation and dialysis.
- Interpersonal contact through sharing items such as razors or toothbrushes, or contact with open sores of a person who has HBV infection.
Hepatitis C is transmitted via direct percutaneous exposure to infectious blood. Mucous membrane exposures to blood can also result in transmission, although this route is less efficient.
HCV is transmitted primarily through:
Less common transmission routes for HCV include:
- Tattooing in unregulated facilities.
- Needles or other sharp instrument injuries.
Clinical features
Symptoms of all types of viral hepatitis are similar and can include one or more of the following:
- Abdominal pain, nausea, and/or vomiting
- Dark urine or clay-colored stools
- Diarrhea (HAV only)
- Loss of appetite
Learn more about signs and symptoms of HAV infection , HBV infection , and HCV infection .
Hepatitis A and hepatitis B are vaccine-preventable. If you suspect a person has been exposed, testing and treatment can prevent complications and interrupt further transmission.
Vaccination
Hepatitis A and hepatitis B vaccines are safe and effective, and they are the best way to prevent HAV and HBV infections. Recommendations for hepatitis A and hepatitis B vaccination include:
Children including:
- All children aged 12–23 months old.
- Unvaccinated children and adolescents aged 2–18 years old.
People at increased risk for HAV infection:
- International travelers.
- Men who have sex with men.
- People who use injection or non-injection drugs.
- People with occupational risk for exposure.
- People who anticipate close personal contact with an international adoptee.
- People experiencing homelessness.
- People in settings that provide services to adults of which a high proportion have risk factors for HAV infection.
People at increased risk for severe disease from HAV infection:
- People with chronic liver disease.
- People with human immunodeficiency virus (HIV) infection.
Other people recommended for vaccination:
- Pregnant people at risk for HAV infection or severe outcome from HAV infection.
- Anyone who requests vaccination.
Vaccination during outbreaks:
- Unvaccinated people in outbreak settings who are at risk for HAV infection or at risk for severe disease from HAV.
- All infants, and unvaccinated children and adolescents aged 18 years and younger.
- All adults aged 19–59 years.
- Adults aged 60 years and older who request vaccination.
- Adults aged 60 years and older with known risk factors for hepatitis B.
Clinical information for hepatitis A and hepatitis B vaccination
There is currently no vaccine available for hepatitis C. Learn more on how to prevent and control the spread of HCV .
Testing, screening, and diagnosis
Testing is the only way to diagnose a viral hepatitis infection.
Serologic tests for acute infection
- For hepatitis A, test for immunoglobulin M (IgM) anti-HAV.
- For hepatitis B, test for hepatitis B surface antigen (HBsAg) plus IgM antibody to hepatitis B core antigen (anti-HBc).
- For hepatitis C, there is no serologic marker for acute infection.
Serologic tests and testing recommendations for chronic infection
Hepatitis A does not progress to chronic infection. Learn more about hepatitis A testing guidelines .
When screening for the first time, tests for chronic infection should include the triple panel test for three HBV seromarkers:
- Hepatitis B surface antigen (HBsAg).
- Antibody to hepatitis B surface antigen (anti-HBs).
- Total antibody to hepatitis B core antigen (total anti-HBc).
For periodic risk-based testing, consider using the triple panel test, or anti-HBc followed (if positive) by HBsAg and anti-HBs.
Universal screening recommendations:
- Screen all adults aged 18 years and older at least once in their lifetime.
- All pregnant people should be tested for HBsAg during every pregnancy, preferably in the first trimester.
- Infants born to HBsAg-positive people should receive postvaccination serologic testing for HBsAg and anti-HBs.
- Test anyone who requests HBV testing regardless of risk because many people may be reluctant to disclose stigmatizing behaviors.
Risk-based testing is recommended for people with a history of risk, regardless of age, if they were susceptible during the period of increased risk, and periodic testing if there is ongoing risk while susceptible, including:
- People born in regions with intermediate and high HBV endemicity (HBsAg prevalence of 2% or higher).
- People born in U.S. not vaccinated as infants whose parents were born in regions with high HBV endemicity (HBsAg prevalence of 2% or higher).
- Household or sexual contacts of people who are HBsAg-positive.
- People who inject or have injected drugs.
- Patients with alanine aminotransferase (ALT) levels (19 IU/L or higher for women, and 30 IU/L or higher for men) of unknown etiology.
- People with end-stage renal disease, including hemodialysis patients.
- People receiving immunosuppressive therapy.
- People with HIV.
- Donors of blood, plasma, organs, tissues, or semen.
- People with a current or a history of sexually transmitted infection.
- People who are currently or were formerly incarcerated.
- People with HCV infection.
Learn more about hepatitis B testing guidelines .
There is no serologic marker for acute HCV infection. Testing for chronic hepatitis C should include:
- Assay for HCV antibody.
- Qualitative and quantitative nucleic acid tests (NAT) to detect and quantify presence of virus (HCV RNA).
Clinicians should initiate hepatitis C testing with an HCV antibody test with reflex to NAT for HCV RNA if the antibody test is positive/reactive. See the complete testing sequence .
Requiring multiple patient visits to collect samples should be discontinued.
Universal screening is recommended for:
- All adults aged 18 years and older at least once in their lifetime.
- All pregnant people during each pregnancy.
- Anyone who requests HCV testing regardless of risk because many people may be reluctant to disclose stigmatizing behaviors.
One-time testing regardless of age or setting prevalence is recommended for:
- People who have ever injected drugs and shared needles, syringes, and/or other equipment, including those who injected once or a few times many years ago.
- People with persistently abnormal ALT levels.
- People who received clotting factor concentrates produced before 1987.
- People who received an organ transplant or a transfusion of blood or blood components before July 1992.
- People who were notified that they received blood from a donor who later tested positive for HCV infection.
- Health care, emergency medical, and public safety personnel after needle sticks, sharps, or mucosal exposures to HCV-positive blood.
- Untested siblings born to same parent.
- Children born to persons with unknown HCV status during pregnancy if birth parent cannot be tested.
Routine periodic testing is recommended for people with ongoing risk factors while risk factors exist, including:
- People with select medical conditions, such as those who have ever received maintenance hemodialysis.
- People who currently inject drugs and/or share needles, syringes, or other drug preparation equipment.
Learn more about hepatitis C testing guidelines .
Treatment and recovery
Treatment for viral hepatitis varies by the type and severity of each infection.
HAV infection is best addressed through supportive care like rest, fluids, a well-balanced diet, and plenty of fluids to relieve symptoms.
Acute HBV infection is best addressed through supportive care like rest, a well-balanced diet, and plenty of fluids to relieve symptoms. Chronic HBV infection should be monitored for signs of liver disease progression and treated with antiviral drugs.
The Infectious Diseases Society of America (IDSA) and the American Association for the Study of Liver Diseases (AASLD) recommend treatment of acute and chronic HCV infection without a waiting period. More than 95% of people with HCV infection can be cured regardless of HCV genotype with 8–12 weeks of oral therapy.
- Hall EW, Bradley H, Barker LK, Lewis K, Shealey J, Valverde E, Sullivan P, Gupta N, Hofmeister MG. Estimating hepatitis C prevalence in the United States, 2017-2020 . Hepatology. 2024 May 13.
Hepatitis is an inflammation of the liver often caused by a virus. Learn about viral hepatitis, statistics, surveillance, resources, populations and impact.
For Everyone
Health care providers, public health.
Overview of Acute Viral Hepatitis
- Symptoms and Signs |
- Diagnosis |
- Treatment |
- Prevention |
- Key Points |
Acute viral hepatitis is diffuse liver inflammation caused by specific hepatotropic viruses that have diverse modes of transmission and epidemiologies. Although acute viral hepatitis can be asymptomatic, a nonspecific viral prodrome is often followed by anorexia, nausea, and often fever or right upper quadrant pain. Jaundice can develop, typically as other symptoms begin to resolve. Most cases resolve spontaneously, but some progress to chronic hepatitis. Occasionally, acute viral hepatitis progresses to acute liver failure (indicating fulminant hepatitis). Diagnosis is by liver tests and serologic tests to identify the virus. Good hygiene and universal precautions can prevent acute viral hepatitis. Depending on the specific virus, preexposure and postexposure prophylaxis may be possible using vaccines or serum globulins. Treatment is usually supportive.
(See also Causes of Hepatitis and Neonatal Hepatitis B Virus Infection .)
Acute viral hepatitis is a common, worldwide disease that has different causes; each type shares clinical, biochemical, and morphologic features. The term acute viral hepatitis often refers to infection of the liver by one of the hepatitis viruses. Other viruses (eg, Epstein-Barr virus , yellow fever virus , cytomegalovirus ) can also cause acute viral hepatitis but less commonly.
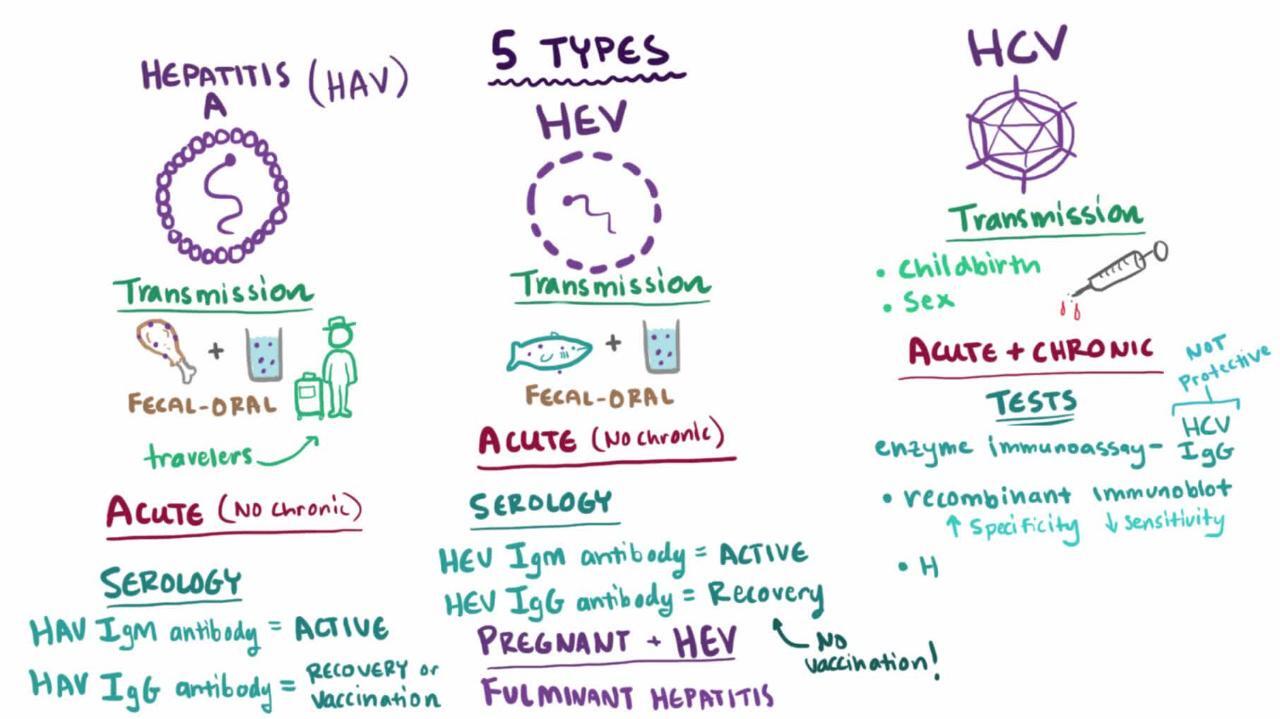
Etiology of Acute Viral Hepatitis
At least 5 specific viruses appear to be responsible (see table Characteristics of Hepatitis Viruses ) for acute viral hepatitis:
Hepatitis A (HAV)
Hepatitis B (HBV)
Hepatitis C (HCV)
Hepatitis D (HDV)
Hepatitis E (HEV)
Other unidentified viruses probably also cause acute viral hepatitis.
Characteristics of Hepatitis Viruses
Symptoms and signs of acute viral hepatitis.
Some manifestations of acute hepatitis are virus-specific (see discussions of individual hepatitis viruses) and some patients are asymptomatic, but in general, acute infection tends to develop in predictable phases:
Incubation period: The virus multiplies and spreads without causing symptoms (see table Characteristics of Hepatitis Viruses ).
Prodromal (pre-icteric) phase: Nonspecific symptoms occur; they include profound anorexia, malaise, nausea and vomiting, a newly developed distaste for cigarettes (in smokers), and often fever or right upper quadrant abdominal pain. Urticaria and arthralgias occasionally occur, especially in HBV infection.
Icteric phase: After 3 to 10 days, the urine darkens, followed by jaundice . Systemic symptoms often regress, and patients feel better despite worsening jaundice. The liver is usually enlarged and tender, but the edge of the liver remains soft and smooth. Mild splenomegaly occurs in 15 to 20% of patients. Jaundice usually peaks within 1 to 2 weeks.
Recovery phase: During this 2- to 4-week period, jaundice fades.
Appetite usually returns after the first week of symptoms. Acute viral hepatitis usually resolves spontaneously 4 to 8 weeks after symptom onset.
Anicteric hepatitis (hepatitis without jaundice) occurs more often than icteric hepatitis in patients with HCV infection and in children with HAV infection. It typically manifests as a minor flu-like illness.
Recrudescent hepatitis occurs in a few patients and is characterized by recurrent manifestations during the recovery phase.
Manifestations of cholestasis may develop during the icteric phase (called cholestatic hepatitis) but usually resolve. When they persist, they cause prolonged jaundice, elevated alkaline phosphatase, and pruritus, despite general regression of inflammation.
Diagnosis of Acute Viral Hepatitis
Liver tests (aspartate aminotransferase [AST] and alanine aminotransferase [ALT] elevated out of proportion to alkaline phosphatase, usually with hyperbilirubinemia)
Viral serologic testing
Prothrombin/international normalized ratio (PT/INR) measurement
Initial diagnosis of acute hepatitis
Acute hepatitis must first be differentiated from other disorders that cause similar symptoms. In the prodromal phase, hepatitis mimics various nonspecific viral illnesses and is difficult to diagnose. Anicteric patients suspected of having hepatitis based on risk factors are tested initially with liver tests, including aminotransferases, bilirubin, and alkaline phosphatase. Acute hepatitis often manifests in the icteric phase and so should be differentiated from other disorders causing jaundice (see figure Simplified Diagnostic Approach to Possible Acute Viral Hepatitis ).
Acute hepatitis can usually be differentiated from other causes of jaundice by
Its marked elevations of AST and ALT: Often ≥ 400 IU/L (6.68 microkat/L)
ALT is typically higher than AST, but absolute levels correlate poorly with clinical severity. Values increase early in the prodromal phase, peak before jaundice is maximal, and fall slowly during the recovery phase. Urinary bilirubin usually precedes jaundice. Hyperbilirubinemia in acute hepatitis varies in severity, and fractionation has no clinical value. Alkaline phosphatase is usually only moderately elevated; marked elevation suggests extrahepatic cholestasis and prompts imaging tests (eg, ultrasonography).
Liver biopsy is usually not needed unless the diagnosis is uncertain.
If laboratory results suggest acute hepatitis, particularly if ALT and AST are > 1000 IU/L (16.7 microkat/L), PT/INR is measured to assess liver function.
Manifestations of portosystemic encephalopathy combined with bleeding diathesis or prolongation of INR suggest acute liver failure , indicating fulminant hepatitis .
If acute hepatitis is suspected, efforts are next directed toward identifying its cause. A history of exposure may provide the only clue of drug-induced or toxic hepatitis. The history should also elicit risk factors for viral hepatitis.
Prodromal sore throat and diffuse adenopathy suggest infectious mononucleosis rather than viral hepatitis.
Simplified Diagnostic Approach to Possible Acute Viral Hepatitis
In patients with findings suggesting acute viral hepatitis, the following studies are done to screen for hepatitis viruses A, B, and C:
IgM antibody to HAV (IgM anti-HAV)
Hepatitis B surface antigen (HBsAg)
IgM antibody to hepatitis B core (IgM anti-HBc)
Antibody to HCV (anti-HCV)
Hepatitis C RNA (HCV-RNA) polymerase chain reaction
If any are positive, further serologic testing may be necessary to differentiate acute from past or chronic infection (see tables Hepatitis A Serology , Hepatitis B Serology , and Hepatitis C Serology ).
If serologically confirmed HBV infection is severe, anti-HDV is measured.
If the patient has recently traveled to an endemic area or is immunosuppressed, IgM antibody to HEV (IgM anti-HEV) should be measured if the test is available.
Biopsy is usually unnecessary but, if done, usually reveals similar histopathology regardless of the specific virus:
Patchy cell dropout
Acidophilic hepatocellular necrosis
Mononuclear inflammatory infiltrate
Histologic evidence of regeneration
Preservation of the reticulin framework
HBV infection can occasionally be diagnosed based on the presence of ground-glass hepatocytes (caused by HBsAg-packed cytoplasm) and using special immunologic stains for the viral components. However, these findings are unusual in acute HBV infection and are much more common in chronic HBV infection.
Treatment of Acute Viral Hepatitis
Supportive care
Treatment of acute hepatitis C, partly to prevent transmission to others
No treatments attenuate acute viral hepatitis. Alcohol should be avoided because it can increase liver damage. Restrictions on diet or activity, including commonly prescribed bed rest, have no scientific basis.
Patients with acute HCV infection should be treated with antiviral therapy upon initial diagnosis without awaiting spontaneous resolution in order to prevent transmission to others. Owing to the high efficacy and safety, the same regimens that are recommended for chronic HCV infection are recommended for acute infection ( 1 ).
Viral hepatitis should be reported to the local or state health department.
Treatment reference
1. American Association for the Study of Liver Diseases (AASLD) and Infection Diseases Society of America (IDSA) : HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. Management of Acute HCV Infection. Accessed May 7, 2024.
Prevention of Acute Viral Hepatitis
Because treatments have limited efficacy, prevention of viral hepatitis is very important.
General measures
Good personal hygiene helps prevent transmission, particularly fecal-oral transmission as occurs with HAV and HEV.
Blood and other body fluids (eg, saliva, semen) of patients with acute HBV and HCV infection and stool of patients with HAV infection are considered infectious. Barrier protection is recommended, but isolation of patients does little to prevent spread of HAV and is of no value in HBV or HCV infection.
Posttransfusion infection is minimized by avoiding unnecessary transfusions and by screening all donors for hepatitis B and C. Screening has decreased the incidence of posttransfusion hepatitis B and hepatitis C, which are now extremely rare in the United States.
Immunoprophylaxis
Immunoprophylaxis can involve active immunization using vaccines and passive immunization.
Vaccines for hepatitis A and hepatitis B are available in the United States.
Routine vaccination for hepatitis A and B is recommended in the United States for all children and for adults at high risk (see Adult Immunization Schedule ).
A vaccine for hepatitis E is not available in the United States but is available in China.
No product exists for immunoprophylaxis of HCV or HDV. However, prevention of HBV infection prevents HDV infection. The propensity of HCV for changing its genome hampers vaccine development.
Transmission is the fecal-oral route for hepatitis A and E parenterally or via blood for hepatitis B and C.
Hepatitis B and C, unlike hepatitis A, predispose to chronic hepatitis and liver cancer (if chronic).
Patients with acute viral hepatitis may be anicteric or even asymptomatic.
Do viral serologic testing (IgM anti-HAV, HBsAg, anti-HCV) if clinical findings are consistent with acute viral hepatitis and AST and ALT are elevated out of proportion to alkaline phosphatase.
Treat patients supportively. Treat acute hepatitis C to prevent transmission.
Routine vaccination for hepatitis A and B is recommended in the United States for all children and for adults at high risk.

Copyright © 2024 Merck & Co., Inc., Rahway, NJ, USA and its affiliates. All rights reserved.
- Cookie Preferences

An official website of the United States government
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
- Publications
- Account settings
- Advanced Search
- Journal List
Viral hepatitis: Past, present, and future
Matthew august odenwald, sonali paul.
- Author information
- Article notes
- Copyright and License information
Author contributions: Odenwald MA and Paul S both contributed equally to this work. Both wrote and revised this review article, and both authors approve of the final manuscript.
Corresponding author: Sonali Paul, MD, MS, Assistant Professor, Department of Medicine, Section of Gastroenterology, Hepatology, and Nutrition, Center for Liver Diseases, University of Chicago, 5841 S. Maryland Ave. Rm J-517, MC7120, Chicago, IL 60637, United States. [email protected]
Received 2021 Jun 27; Revised 2022 Mar 4; Issue date 2022 Apr 14.
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Each hepatitis virus—Hepatitis A, B, C, D, E, and G—poses a distinct scenario to the patient and clinician alike. Since the discovery of each virus, extensive knowledge regarding epidemiology, virologic properties, and the natural clinical and immunologic history of acute and chronic infections has been generated. Basic discoveries about host immunologic responses to acute and chronic viral infections, combined with virologic data, has led to vaccines to prevent Hepatitis A, B, and E and highly efficacious antivirals for Hepatitis B and C. These therapeutic breakthroughs are transforming the fields of hepatology, transplant medicine in general, and public and global health. Most notably, there is even an ambitious global effort to eliminate chronic viral hepatitis within the next decade. While attainable, there are many barriers to this goal that are being actively investigated in basic and clinical labs on the local, national, and international scales. Herein, we discuss pertinent clinical information and recent organizational guidelines for each of the individual hepatitis viruses while also synthesizing this information with the latest research to focus on exciting future directions for each virus.
Keywords: Viral Hepatitis, Hepatitis A, Hepatitis B, Hepatitis C, Hepatitis D, Hepatitis E, Hepatitis G
Core Tip: Viral hepatitis encompasses a wide array of clinical diseases—from asymptomatic and self-limited to chronic liver disease to acute liver failure. Extensive historical research has resulted in vaccines to prevent Hepatitis A, B, and E and highly efficacious antivirals for Hepatitis B and C, and these therapeutic breakthroughs are transforming the fields of hepatology, transplant medicine in general, and public and global health. While these breakthroughs are highly promising, there are many barriers to eventually elimination of chronic viral hepatitis. These barriers are being actively investigated, and we discuss ongoing research in the historical context of viral hepatitis research.
INTRODUCTION
In this review of viral hepatitis infections, we discuss the pertinent clinical information and recent organizational guidelines for each of the individual hepatitis viruses while also synthesizing this information with the latest research to focus on exciting future directions for each virus.
HEPATITIS A
Hepatitis a.
The Hepatitis A Virus (HAV) is a single stranded, non-enveloped ribonucleic acid (RNA) molecule. HAV is a member of the Picornaviridae family and the Hepatovirus genus that is transmitted primarily through a fecal-oral route via person-to-person contact or ingestion of contaminated food or water[ 1 , 2 ]. Globally, serologic evidence of prior infection is quite high, but prevalence has high geographic and demographic variability[ 3 ]. Acute HAV infection characteristically causes a self-limited illness. However, cases of fulminant liver failure have been reported with advanced age being the greatest risk factor for symptomatic disease[ 4 ]. Treatment is primarily preventative with vaccination prior to possible exposures, and both vaccination and HAV immunoglobulin to confer both active and passive immunity after exposure[ 5 ].
Epidemiology
An estimated 1.4 million cases of hepatitis A occur globally each year[ 6 ]. Estimates of disease prevalence vary regionally and are highly dependent on socioeconomic status and access to clean water. In developing countries with poor sanitation, there is nearly 100% seropositivity for HAV immunoglobulin G (IgG). In these countries, it is presumed that most children are infected at very early ages, when minimal symptoms develop and therefore clinical presentation of HAV infection is very rare[ 3 ]. In wealthy nations, including the United States, HAV IgG seropositivity rates are much lower[ 7 ]. Seropositivity increased with age and was lower among United States-born residents compared to immigrants[ 8 ]. Since the introduction of the HAV vaccine in 1996, new cases of HAV have declined by over 90% in the United States despite relatively low rates of HAV vaccination[ 9 ]. International travel to endemic areas or person-to-person contact with an infected person are the main risk factors for HAV.
Natural History of Infection and Clinical Course
The primary route of HAV infection is the fecal-oral route from contaminated food and water. This is evidenced by detection of HAV RNA in stool during the incubation period and for up to 4-5 mo after the onset of symptoms[ 10 ]. In small children, the infection is largely asymptomatic with fewer than 10% of children < 6 years old developing jaundice with HAV infection, and the only evidence of infection is serologic presence of anti-HAV antibodies. In older children and adults, HAV infection follows a characteristic pattern[ 5 ]. Infection is followed by an average 28-d incubation period where the virus actively replicates in hepatocytes. HAV itself is not thought to be directly hepatotoxic as there is no laboratory or clinical evidence of liver damage during the incubation period, and HAV can be propagated in vitro without any evidence of cytopathology[ 11 ]. After the incubation period, however, there is immune-mediated damage to the hepatocytes that results in non-specific symptoms of fever, malaise, fatigue, and loss of appetite followed by jaundice in approximately 70% of patients[ 12 ]. The majority of patients (approximately 60%) have a full recovery within 2 mo. Of the patients who do not fully recover within 2 mo, some develop prolonged cholestasis and others have relapsing disease with 2 or more bouts within a 6-10 wk period driven primarily by viral shedding within the stool and reinfection. The overwhelming majority (nearly 100%) of these patients fully recover within 6 mo of disease onset, and there is no increase in mortality with any of these disease presentations[ 9 ].
Fulminant HAV is associated with low HAV RNA titers and high bilirubin levels, likely related to a robust host immune response reducing HAV viral load and resulting in significant hepatocyte damage[ 4 ]. Acute liver failure occurs in less than 1% of cases of HAV infection[ 4 ]. It is more common among patients with advance age (> 75 years old), underlying liver disease, or chronic kidney disease. The incidence of fulminant HAV in the United States has decreased dramatically from 1990 to 2005[ 13 ]. Data from the United States Acute Liver Failure Study Group (ALFSG) showed that the proportion of ALFSG cases due to HAV was low (29 of 925 patients, 3.1%), and of these patients, 55% recovered, 31% received transplant, and 14% died[ 13 ]. In 2006, the ALFSG study group designed a prognostic model based on clinical features at presentation [alanine transaminase (ALT) < 2600 IU/L, creatinine > 2.0 mg/dL, intubation, and vasopressors] that predict the likelihood of death and need for transplant with high accuracy[ 13 ]. Subsequently, a refined scoring system was derived from a cohort of 294 Korean patients with fulminant hepatitis A to predict the likelihood of death or need for liver transplant[ 14 ]. This scoring system takes multiple objective values (age, international normalized ratio, bilirubin, ammonia, creatinine, and hemoglobin) at the time of HAV-associated ALF into account, and compared to the ALFSG study group, this new model better predicted the likelihood of death or need for transplantation in both the Korean discovery cohort and international validation cohorts[ 14 ]. These scoring systems are useful in determining the level of care that a patient with acute HAV infection should receive. Nevertheless, there is an unusually high rate of recovery for HAV-related acute liver failure, and given this, auxiliary transplantation and artificial liver devices have been proposed as therapeutic bridges to native liver recovery and regeneration[ 15 ]. However, these are not commonly used in clinical practice.
Prevention, Diagnosis, and Treatment
There are currently 4 inactivated HAV vaccines available, all with similar efficacy and side effect profiles. However, widespread vaccination programs are not currently universal[ 16 ]. In fact, the World Health Organization (WHO) recommends that large-scale efforts should not be undertaken in highly endemic areas where nearly 100% of children contract HAV early in life and are asymptomatic[ 16 ]. On the other hand, in regions with lower rates of disease and higher acute infection rates later in life (when it is more likely to be symptomatic and result in increased healthcare costs) the WHO recommends either targeted vaccination of high-risk groups (in very low prevalence areas) or universal vaccination programs (in intermediate endemic areas)[ 16 ]. Given recent outbreaks and increases in the number of cases reported in the United States each year, the United States Centers for Disease Control (CDC) now recommends vaccination of all children > 1 year old in addition to the traditional at-risk groups[ 17 ].
Clinically, HAV infection is indistinguishable from other forms of acute viral hepatitis and diagnosis relies on serologies. An acute infection is defined by the presence of anti-HAV-immunoglobulin M (IgM) antibodies, which are present within a couple of weeks of exposure and at the onset of symptoms. Anti-HAV-IgG antibodies are also present at the onset of symptoms. While the anti-HAV-IgM titer decreases over time and is generally undetectable after 1 year of exposure, the IgG antibody is present for life and confers lifelong immunity. HAV RNA can be found in various bodily secretions and excretions, which can determine infectivity but levels are not currently used clinically.
Treatment for HAV is largely supportive with spontaneous recovery in the overwhelming majority of patients. While there is no anti-viral treatment for HAV, some studies have investigated post-exposure prophylaxis with both active immunity with HAV vaccination and conferring passive immunity through HAV immunoglobulin infusions. The most comprehensive study came in 2007 when Victor et al [ 18 ] performed a randomized control trial comparing HAV vaccination to HAV immunoglobulin in 1090 household contacts aged (2 to 40) of HAV patients. Both groups had low rates of hepatitis A, and the study’s noninferiority criteria were met[ 18 ]. As such, the United States CDC recommends either HAV vaccine or HAV immunoglobulin for post-exposure prophylaxis within 2 wk of exposure[ 19 ]; however, the HAV vaccine does have an advantage over immunoglobulin, including active immunity and longer duration of action.
In summary, while effective HAV vaccines are available, the available data support the current practice of targeted vaccination in areas where patients who are more prone to more severe symptoms from HAV are more likely to be exposed rather than vaccinating all individuals in endemic areas. For those who are exposed, it will be interesting to see if further improvements can be made to already good predictive models to determine the clinical trajectory of patients with acute, fulminant HAV to determine whether liver transplant will be needed. Finally, we will be eagerly watching for further data on currently investigational liver support devices, which hold promise to provide supportive care through fulminant HAV and obviate the need for liver transplantation.
HEPATITIS B
Hepatitis B infection is caused by the Hepatitis B Virus (HBV), a deoxyribonucleic acid (DNA) virus belonging to the Hepadnaviridae family and the Orthohepadnavirus genus[ 20 ]. It is transmitted via exposure to infected blood or bodily fluids, most commonly from intravenous drug use, sexual contact, or vertical transmission from mother to child[ 20 ]. The burden of HBV is declining in the developed world due to vaccination[ 21 ], but HBV prevalence is still quite high in endemic areas primarily due to vertical transmission between mother and child and early life exposures[ 22 ]. The age of HBV infection is the principal factor determining the course of disease; the overwhelming majority of perinatally infected patients develop chronic hepatitis B whereas the majority of adults who are infected readily clear the virus[ 23 ]. Antiviral medications can stop viral replication and subsequent liver damage. While no available treatments can clear HBV infection, there are exciting investigational agents that may provide therapeutic benefit in the future[ 24 ]. Moreover, there is a broad global health effort to eliminate HBV via a combination of aggressive vaccination, diagnostic, and treatment programs[ 25 ].
In 2006, it was estimated that 2 billion people had been infected with HBV and that 360 million people were living with chronic hepatitis B worldwide. There is geographic variation in hepatitis B prevalence. Endemic regions like Southeast Asia, Sub-Saharan Africa, and parts of South America have prevalence rates greater than 8% compared to 2% in non-endemic areas, including the majority of North America[ 26 ]. Routes of transmission differ between endemic and non-endemic areas and determine the course of HBV infection. In endemic areas, vertical transmission between mother and child and horizontal transmission among young children are the most common routes of HBV infection, but in non-endemic areas, intravenous drug use and sexual transmission in adults are the predominant modes of infection[ 27 ]. Exposure to HBV within the first six months of life confers a nearly 90% risk of developing a chronic infection due to immunologic tolerance, which decreases to approximately 50% risk if exposed before the age of 6[ 27 ]. Acutely infected adults with intact immune systems, however, spontaneously clear HBV infection in a remarkable 95% of cases. Taken together, these data indicate that the majority of chronic HBV in the world is within endemic areas. In line with this, recent studies have shown that more than 90% of cases of chronic HBV in the United States are in immigrants from endemic areas[ 28 ]. While the incidence of acute HBV is declining in the United States due to vaccination, blood product screening, and perinatal screening, the incidence of chronic HBV is increasing due to changing immigration patterns and increasing immigration from endemic areas[ 28 ]. A recent meta-analysis assessed the prevalence of hepatitis B surface antigen (HBsAg) in hemodialysis patients in the Middle East found a 4.4% positivity rate, which is decreasing over time[ 29 ].
Hepatitis B is a small DNA virus with 10 known genotypes. The enveloped hepatitis B virus is recognized via the HBsAg and enters the cell via receptor-mediated endocytosis[ 20 ]. Upon entry into the cell, HBV is uncoated and undergoes repair of the single-stranded DNA to either integrate into the host genome or form covalently closed circular DNA (cccDNA), both of which serve as templates for transcription and translation[ 20 ]. The cccDNA persists in hepatocytes even after other signs of declining virus activity, including HBsAg loss, and is the main cause of HBV persistence despite antiviral treatment. The 3.2kb genome has 4 open reading frames that encode for the (1) Core gene (important for viral packaging and production of e-antigen (eAg); (2) surface gene (encodes surface proteins); (3) X-gene (which maintains expression of cccDNA); and (4) polymerase gene (encodes multiple proteins important for DNA replication, including a reverse transcriptase and polymerase)[ 24 ]. Once transcribed from cccDNA, immature RNA molecules are packaged into nucleocapsids that can either be recycled to the nucleus or further packaged and trafficked to budding sites in a HBsAg-dependent manner.
HBV is not directly cytotoxic but instead the clinical course of HBV is determined by the intensity of the immune response[ 30 , 31 ]. Acute HBV infection is a subclinical illness in approximately 2/3 of the cases while the other 1/3 develop symptomatic hepatitis and 1% develop acute liver failure with survival rates of only 20%. Regardless of the initial clinical manifestation, with an intact immune system, the majority of HBV is rapidly cleared, and understanding the modes of natural clearance are important for developing new treatment strategies.
The innate immune system is classically thought of as the initial line of defense against pathogens: It uses non-specific defenses including proinflammatory cytokines, interferons, and natural killer (NK) cells to keep the virus under reasonable control[ 31 ]. The adaptive immune system is fine-tuned to more specifically fight the pathogen. In hepatitis B, however, the line between these two arms of the immune system are blurred. In acute HBV infection, the innate immune system has a relatively weak release of the prototypical cytokines and weak induction of interferon and interferon-stimulated genes[ 32 ]. In fact, HBV has been shown to actively suppress interferon release and inhibit with interferon functions[ 33 ]. In one study of 21 patients with acute HBV infection, these patients had an increase in the anti-inflammatory cytokine interleukin (IL)-10 while there was no change in interferon levels from baseline[ 34 ]. Increases in IL-10 were accompanied by a decrease in NK cell activation and attenuated HBV-specific CD4 + and CD8 + T-cell responses[ 34 ]. Moreover, there is a decrease in NK cell function in clearing the initial infection. However, non-traditional roles for NK cells, including regulating the adaptive T-cell response, and the non-classical NK T-cells (NKT cells) can clear HBV without the help of CD4 + or CD8 + T-cells. These defects in the innate immune response to HBV infection are likely the culprit for the relatively low rate of symptomatic hepatitis in the acute phase and have led to HBV being considered a “stealth virus”[ 35 ].
Adaptive immunity plays a crucial role in clearance of acute HBV infection. Classically, CD8 + T-cells selectively eliminate virus-infected cells by recognizing short viral epitopes on infected cells, and HBV-specific CD8 + T-cells are an integral component of natural HBV control[ 31 ]. This was suggested by a study of 23 patients with acute, self-limited HBV[ 36 ]. Interestingly, the highest frequency of HBV-specific CD8 + T-cells correlated with the clinically acute phase of infection[ 36 ]. Causation was more convincingly demonstrated with an experimental chimpanzee model with selective depletion of either CD4 + T-cells or CD8 + T-cells prior to acute HBV infection[ 37 ]. Chimpanzees with depletion of CD8 + T-cells had delayed viral clearance, and viral clearance did not occur until the reappearance of CD8 + T-cells[ 37 ]. Repopulation of CD8 + T-cells and viral clearance also coincided with interferon expression. Depletion of CD4 + T-cells did not have any appreciable effect on viral clearance compared to controls[ 37 ].
Patients who are unable to mount this initial immune response fail to clear the virus and progress to chronic HBV. Immunologic hallmarks of chronic hepatitis B infection include numerical and functional deficiency of HBV-specific CD8 + T-cells as well as decreased B-cell function[ 38 - 40 ]. Given their crucial role in clearing acute HBV infection, augmenting the number and function of CD8 + T-cells is of great interest in therapeutic development.
Chronic hepatitis B has four distinct phases: the immune tolerant phase, immune active phase, inactive carrier phase, and reactivation. In the immune tolerant phase, there are high HBV viral loads without lab evidence of liver inflammation. Immune active phase is evidenced by lower viral loads with elevated transaminases. If untreated, patients in the immune active phase have a very high chance (approximately 20%) of progressing to chronic liver disease with cirrhosis and hepatocellular carcinoma (HCC) in approximately 25%-30% of patients with the presence of active viral replication and necroinflammatory liver disease being predictors of disease progression. In the absence of antiviral therapy, patients with HBsAg-positive cirrhosis have an 84% 5-year survival when compensated but a bleak 14% 5-year survival rate after the initial decompensation event[ 41 ]. While current antivirals can help improve liver histology, decrease hepatic decompensation, and improve long-term survival, achieving a functional cure ( i.e. , HBsAg loss) is an uncommon event with unknown predictive factors[ 42 , 43 ]. In the inactive carrier state, patients have normalization of transaminases, undetectable HBV virus levels, and in some patients, fibrosis improvement[ 44 ]. However, these patients can reactivate either due to loss of immune control whether spontaneously or induced by immunosuppressive therapies[ 45 ]. Despite available treatments, the burden of chronic HBV is still very high and is estimated to account for 700000 deaths each year from decompensated cirrhosis and HCC[ 46 ].
As discussed above, vertical and horizontal transmission in childhood are responsible for the majority of chronic HBV infections. HBV can be prevented with administration of an effective HBV vaccine, which has been available since the 1980s. This was demonstrated with mass childhood vaccination programs in the endemic area of Taiwan that started in 1984[ 47 ]. Seroprevalence of HBV was tested 10 years after the introduction of mass vaccination programs and showed marked declines in the childhood presence of HBsAg (from 9.8% to 1.3%) and rising rates of immunity as marked by hepatitis B surface antibody (HBsAb) (from 23% to 79%)[ 47 ]. Importantly, a subsequent study showed that universal childhood HBV vaccination was also linked to reduced incidence of childhood HCC and HCC-associated mortality[ 48 ]. A similar aggressive vaccination program for Native Alaskans was conducted in newborns and young-children and was linked to elimination of acute, symptomatic HBV infections and HCC[ 49 ]. Additionally, despite a growing population, the number of children identified with HBsAg fell from 697 to 2 after initiation of this vaccination program[ 49 ]. The effect of vaccination has also been studied in infants at highest risk of vertical transmission—those born to HBeAg positive, HBsAg carrier mothers[ 50 ]. In combination with hepatitis B immunoglobulin (HBIG), neonatal vaccination for HBV was extremely efficacious. Persistent HBsAg was found in only 6% of the infants receiving both vaccination and HBIG compared to 88% of those receiving placebo[ 50 ]. With this marked efficacy in mind, the World Health Organization has recommended universal birth dose vaccination against HBV. However, despite the demonstrated efficacy more than 35 years ago, this has still not occurred in the majority of endemic countries. Expanding HBV vaccination is therefore the focus of many ongoing global health efforts to eliminate HBV[ 25 ].
Available antiviral medications against HBV are interferon-based regimens or nucleotide/nucleoside reverse transcriptase inhibitors (NRTIs), including entecavir, tenofovir, lamivudine, adefovir, and telbivudine[ 42 ]. Interferon-based therapies have both immune stimulating and antiviral effects and have higher potency than NRTIs; however, it is used much less frequently because of its adverse effects. The first generation NRTIs (lamivudine, adefovir, and telbivudine) suffered from low-barriers to viral resistance and have been replaced by second generation NRTIs (entecavir and tenofovir) in clinical practice[ 42 ]. When used alone, NRTIs can achieve HBV DNA negativity in 70%-85% of patients and e-antigen seroconversion in 20%-25% of patients after 1 year of treatment with even further benefit after 3 years[ 51 ]. However, even patients with seroconversion have high rates of HBV relapse upon withdrawal of medication (approximately 30% by 5 years), which has led to prolonged therapy even after e-antigen seroconversion to decrease the risk of reactivation[ 52 ]. While many patients are treated with lifelong NRTIs with minimal side effects, even the simplest treatment algorithms require physician visits, lab draws, and HCC screening every 6 mo. This is costly and inconvenient, which can lead to non-adherence to care for a variety of reasons, especially in many resource-limited endemic countries. Indeed, worldwide, many patients that guidelines would suggest to be on NRTI therapy do not actually receive treatment[ 25 ]. Multiple approaches to remove this barrier to receiving care are being investigated and include determining if and when therapy can safely be withdrawn and combining NRTI therapy with other therapies to improve durable, off-treatment responses. In retrospective analyses of patients who have discontinued NRTI, it appears safe to stop therapy if patients do not have HBeAg, have very low levels of HBsAg, and have minimal fibrosis. However, flares can still occur and HCC screening is still required, and therefore close follow up is still warranted, suggesting that this approach may not be feasible on a global scale[ 53 ]. NRTI withdrawal appears to be more feasible in non-cirrhotic, HBeAg negative patients with low HBV DNA titers if NRTI is combined with interferon therapy; however, this too was done in a highly regimented clinical trial setting with relatively short-term follow up[ 54 ]. At the moment, this approach is only experimental, and real-world experience with longer term data to determine HBV relapse rates and HCC occurrence has yet to be seen.
In sum, the most promising method for prevention of primary HBV infections is early, universal vaccination, and we are hopeful that aggressive vaccination campaigns already underway will substantially reduce the burden of HBV in the coming decades. Fortunately, highly active antivirals are capable of controlling the virus and reducing the burden of advanced liver disease from HBV; however, there remains no cure for HBV. In the next section, we discuss many exciting experimental approaches aimed at curing HBV via multiple different mechanisms.
Future Therapies Under Investigation
The high rate of HBV relapse after NRTI withdrawal reflects the persistence of cccDNA and integrated HBV DNA. Multiple drugs in development target components of the HBV lifecycle as well as the immune response to HBV infection. Future approaches to achieving a cure for hepatitis B will likely exploit all three pathways. That is, (1) Use of existing potent antivirals in combination with; (2) novel, direct acting antivirals; and (3) immunomodulators that enhance clearance of cells that harbor HBV DNA[ 24 ]. Novel direct-acting antiviral therapies under investigation include gene editing and suppression via clustered regularly interspaced short palindromic repeats (CRISPR-CAS-9) technology and gene suppression via silencing RNA (siRNA) and antisense oligonucleotides (ASOs)[ 24 ].
The CRISPR approach has been used with guide RNAs designed to target cccDNA components (core, polymerase, or X open reading frames)[ 55 ]. This study demonstrated the ability of CRISPR technology to directly cleave cccDNA and significantly reduce both HBV RNA titers and HBsAg concentrations in vitro with both transient transfection and sustained expression. In a mouse model of HBV infection, simultaneous delivery of HBV and guide RNA was also shown to result in decreased HBV viral titers and HBsAg levels[ 55 ]. Given that both integrated HBV DNA and cccDNA can contribute to HBV persistence, another group successfully used CRISPR to excise a full-length integrated DNA fragment while simultaneously disrupting cccDNA in a cell line that stably expressed HBV DNA[ 56 ]. All measures of HBV chronicity were undetectable for 12 mo after this therapy[ 56 ]. While this is promising, CRISPR-Cas9 targets the integrated DNA by inducing double stranded DNA breaks within the host genome, which has the potential to have detrimental off-target effects including host genome rearrangement. More targeted approaches with CRISPR-Cas-9 “nickases” to mediate only single strand nicks and base-editing to introduce mutations have also been employed[ 57 ]. A more targeted approach with CRISPR-Cas9-mediated base-editing to introduce mutations has also been attempted in vitro . The introduction of either missense or nonsense mutations resulted in inactivation of both integrated and cccDNA with associated reductions in HBV viral titers, HBsAg, and reductions in both surface and polymerase proteins[ 57 ]. Moreover, CRISPR-Cas-9-mediated knockout of HBsAg in an HCC cell line reduced proliferation in vitro and the ability of these cells to form tumors in mice, suggesting therapeutic potential for HBV-associated HCC as well[ 58 ]. Together, these data suggest that CRISPR-Cas9 technology has the potential to promote a true HBV cure.
Alternative approaches that directly target the hepatitis B virus include targeting the RNA products of both cccDNA and integrated HBV transcription via specific siRNA or ASOs. Both siRNA and ASOs are short RNA sequences that are complementary to the viral mRNA and therefore generate double stranded RNA that is targeted for degradation via dicer or RNase-H, respectively[ 59 ]. With multiple siRNA or ASOs administered in each dose, this approach has the potential to target multiple mRNA products simultaneously[ 59 ].
One siRNA molecule, ARC-520, is designed to target the open reading frame of HBV X but overlaps with all cccDNA transcripts and therefore has the potential to target all cccDNA transcripts for degradation. ARC-520 was shown to be safe in healthy volunteers[ 60 ], and in a phase II study, ARC-520 resulted in significant reduction in HBsAg production in patients that were NRTI-naïve or had HBeAg[ 61 ]. The difference in response to RNA interference (RNAi) between HBeAg+ and HBeAg- patients was investigated using chimpanzees and determined to be due to the presence of integrated DNA in addition to cccDNA[ 61 ]. In follow up studies of ARC-520 activity against chronic HBV in NRTI-experienced patients, four monthly doses of the siRNA resulted in dose-dependent reduction in HBsAg concentrations regardless of HBeAg status. The reductions were only modest (approximately 0.4 log reduction from baseline), possibly resulting from the presence of integrated DNA that is not targeted by this particular siRNA[ 62 ]. Additional siRNAs are currently under development and preliminary data from one siRNA that uses a N-Acetylgalactosamine ligand to target the siRNA to the liver suggests improved HBsAg reduction (approximately 1.75 log reduction) with dosing every 4 wk[ 63 ].
The potential of ASO in treating chronic HBV was recently demonstrated in preclinical in vitro and in vivo mouse models[ 64 ]. A target second-generation ASO that was complementary to reference sequences in HBV genotypes A-H was identified as an effective ASO using in vitro models. This ASO reduced HBV DNA expression, replication, viremia, and HBsAg and HBeAg production in multiple HBV genotypes[ 64 ]. Importantly, the ASO used had no interference with the anti-viral activity of NRTIs when given simultaneously, suggesting that combination with existing therapy is feasible[ 64 ]. An HBV-targeted ASO was recently shown to be safe in escalating doses in healthy human subjects[ 65 ], and subsequently shown to have excellent antiviral activity in patients with chronic hepatitis B with reduction in HBsAg regardless of concurrent therapy after 29 d[ 66 ]. In addition to the longer-term follow up that is underway, it will be important to determine if there can be a further decline in measures of HBV infection if combined with additional ASOs or if sequence variations between individuals can be accounted for with use of multiple ASOs simultaneously[ 67 ]. Moreover, further modifications to ASOs are currently in development to improve delivery to hepatocytes[ 67 ].
Immunomodulation aims to rectify the relative functional and numerical deficiency of CD8 + T-cells that is present in patients with chronic hepatitis B infections. This can be achieved either by augmenting the function of existing CD8 + T-cells, creating a new source of CD8 + T-cells, or immune mobilization[ 24 , 31 , 68 , 69 ]. One hallmark of CD8 + T-cells in chronic HBV is overexpression of inhibitory molecules, among them programmed cell death protein-1 (PD-1). This has led to the hypothesis that PD-1 inhibition may increase recruitment of T-cells to help clear chronic hepatitis B. Efficacy of a single dose of the PD-1 inhibitor nivolumab was tested in patients who already had viral suppression and were HBeAg negative and demonstrated a modest further reduction in HBsAg production at 12 wk (average reduction was 0.3 log reduction) with only 1 patient achieving loss of HBsAg production in the study period[ 70 ]. While these data demonstrate some additive benefit of immunotherapy with currently available antivirals, it is notable that this study specifically excluded patients with advanced fibrosis leaving open the possibility that patients with cirrhosis may not be ideal candidates for this approach. Moreover, given extremely low number of HBV-specific CD8 + T-cells in chronic hepatitis B, it is unlikely that recruitment of autologous T-cells alone will provide a durable cure.
This realization has led to efforts to generate new functional pools of effector T-cells by engineering large numbers of HBV-specific T-cells using chimeric antigen receptor (CAR) and T-cell receptor (TCR) technology. This approach has been successfully employed in mouse models of chronic HBV. One group isolated CD8 + T-cells from mice and engineered them to express CAR that bind to HBV envelope proteins prior to transferring them into HBV transgenic mice[ 71 ]. These adoptively transferred HBV-specific T-cells engrafted, expanded, honed to the liver, reduced HBV replication, and caused only transient liver damage[ 71 ]. Similar success was achieved in HBV-infected humanized mice using human T-cells that were engineered to express an HBV-specific T-cell receptor[ 72 ]. Further, simultaneous treatment with the TCR-engineered cells and the HBV entry inhibitor myrcludex led to long-term control of HBV infection with limited liver injury[ 72 ].
A third approach has recently been described and acts as a hybrid between the above described methods of recruitment autologous T-cells and engineering larger pools of T-cells[ 69 ]. This approach uses immune-mobilizing monoclonal T-cell receptors against the virus (immTAV), which are soluble bispecific proteins that bind to both the TCR, which specifically recognizes the HBV viral peptide-human leukocyte antigen complex, and to CD3, which recognizes non-specific T-cells. Using an HBV envelope protein-specific immTAV, one group demonstrated that this approach can redirect polyclonal T-cells to destroy hepatocytes that are either infected with HBV or have integrated HBV DNA[ 69 ].
Despite the success of these immunotherapy approaches in cell culture and animal models, these have not yet been translated into human trials. Investigators are likely to proceed with caution given that these cytotoxic T-cells are targeting infected tissue within an essential organ and any exaggerated or off-target effect has the potential to induce irreparable liver damage and prove fatal. One possible way to minimize this chance is to minimize the portion of hepatocytes harboring HBV infection with existing antivirals. It is therefore likely that this approach will be most useful after existing or novel antiviral medications deplete significant portions of HBV DNA. Enhancing CD8 + T-cell function will therefore result in destruction of the few remaining infected hepatocytes to allow for a cure.
HEPATITIS C
Hepatitis C infection is caused by the Hepatitis C virus (HCV), a single stranded RNA virus. HCV is a member of the Flaviviridae family and Hepacivirus genus that is transmitted primarily through direct bloodstream inoculation[ 73 , 74 ]. HCV successfully evades the immune system to cause a chronic hepatitis in the majority of cases, which often leads to advanced fibrosis and cirrhosis if untreated[ 73 ]. While there are no effective vaccines for HCV prevention, with the advent of direct-acting antivirals (DAA) for the treatment of HCV, HCV can be easily cured in the overwhelming majority of cases. This has now led to ambitious global efforts to eliminate HCV[ 75 ]. Additionally, DAAs have led to the exciting possibility of transplanting organs from HCV-positive donors, which has the potential to greatly expand the organ donor pool and increase the availability of scarce resources.
HCV is estimated to effect approximately 3% of the population worldwide, which translates into nearly 200 million cases worldwide. There is a wide geographic variation in disease prevalence with rates of approximately 1.5% in the United States to nearly 15% in Egypt and up to 50% in certain age groups in Egypt[ 74 , 76 , 77 ]. The unusually high rate of HCV positivity in Egypt has been traced to campaigns to administer parenteral anti-schistosomiasis treatment with inadequate needle sterilization in the 1950-1980s and subsequent spread with blood transfusions and medical and dental procedures[ 78 ]. More recently, a large effort to screen and treat Egyptians for HCV was undertaken and successfully screened 49.6 million people for HCV and showed a lower seroprevalence than previous estimates after large treatment efforts, now at 4.61%[ 79 ]. Remarkably, after DAA treatment 98.8% of patients with a known treatment outcome had achieved sustained viral response, suggesting an even lower disease prevalence in Egypt now[ 79 ].
The estimates of prevalence in the United States are based on the National Health and Nutrition Examination Survey (NHANES) database, which surveys a representative sample of 5000 adults annually; however, it is notable that this survey does not include homeless or incarcerated individuals, where HCV prevalence is estimated to be significantly higher, with weighted prevalence of 23.1% in the incarcerated population and 32.1% in the homeless[ 80 ]. The estimates in the United States are therefore likely to underestimate the true disease burden with one study calculating that NHANES underestimates the number of HCV infections in the US by approximately 1 million[ 80 ]. Moreover, in recent years, the opioid epidemic has led to increasing rates of HCV cases and a shifting demographic, now with rates of infection rising fastest in young people while rates in the baby boomer generation fall due to screening efforts and treatment[ 81 ].
Hepatitis C transmission occurs with parenteral exposures, most commonly from intravenous drug use and contaminated blood transfusions with a notable exception in Egypt, as discussed above. A case control series from 1997 surveyed both 2316 HCV-positive and 2316 HCV-negative blood donors from the United States for lifestyle and socioeconomic factors to elucidate possible mechanisms of HCV exposure and transmission and to assist with finding populations that may benefit from closer screening[ 82 ]. They found that the strongest risk factors for HCV-positive individuals were intravenous (IV) drug use (OR 49.6), blood transfusion, (OR 10.9), and sex with an IV drug user (OR 6.3)[ 82 ]. Since 1992, the United States has enacted universal HCV screening of donated blood, which has dramatically reduced the risk of HCV transmission via blood transfusions[ 83 ]. HCV can survive on an unsterilized needle for many days depending on the temperature and inoculum[ 84 ]. There are an estimated 11-21 million IV drug users worldwide[ 85 ], with rates rising in many countries, most notably with the opioid epidemic in the United States[ 81 ]. This has already led to rates of HCV infection rising sharply since 2010[ 86 ] and is likely to continue to do so until the opioid epidemic is under better control.
Nearly all cases of acute hepatitis C are asymptomatic, and fulminant hepatitis C has been reported only in case reports[ 87 ]. The lack of symptoms in early infections has precluded identification of patients for large studies to fully characterize this phase of the disease. Studies of transfusion-associated infections have been able to identify some patients with acute, transfusion-related HCV infections[ 88 ]. These studies have identified that HCV RNA rises rapidly after infection followed by a rise in ALT in 1-3 mo, indicating hepatocyte damage. The latent period (defined as the period prior to ALT elevations) is inversely proportional to the donor HCV viral load and is shorter in patients with clinically overt disease[ 88 ]. Importantly, these studies have helped identify host factors that assist with HCV clearance and those that make hosts susceptible to developing chronic disease. More recently, the factors that mediate this rapid immune response are being used in the hopes of developing a hepatitis C vaccine.
One such study analyzed women who were infected with HCV when prophylactically treated with Rh o (D) immunoglobulin and found that spontaneous viral clearance is strongly associated with genetic polymorphisms near the interleukin 28B ( IL28B ) gene, which encodes interferon (IFN)-λ-3[ 89 ]. Moreover, women who did not have the favorable IL28B polymorphism had increased chance of viral clearance if they developed jaundice in the acute phase of infection[ 89 ]. This finding has been confirmed in additional studies, and one study followed 632 patients with acute HCV and found that 25% of patients cleared acute HCV with clearance being more likely if patients were female, had the favorable IL28B genotype (C/C), or were infected with HCV genotype 1[ 90 ]. This large study suggests that HCV becomes a chronic infection in approximately 75% of acutely infected individuals, which is in line with widely quoted estimates of 50%-85% chronicity rates[ 90 ].
Chronic HCV is also largely asymptomatic prior to the development of advanced fibrosis, and cirrhosis is estimated to occur in 16% of patients within 20 years of HCV infection[ 91 ]. Factors that contribute to chronic HCV progressing to cirrhosis include advanced age, concurrent HBV, ongoing alcohol use, immunocompromised states, and risk factors for non-alcoholic steatosis including obesity and insulin resistance[ 92 ]. Once a patient has cirrhosis, they are at higher risk for hepatic decompensation and the development of HCC. While HCV-associated HCC can develop in non-cirrhotic livers[ 93 ], the risk is much higher in patients with cirrhosis and additional risk is conferred by many of the factors associated with progression to cirrhosis in the first place, including age, alcohol use, and male sex[ 92 ].
Diagnosis and Treatment
As briefly discussed above, Hepatitis C is almost universally a chronic, asymptomatic disease until it ultimately causes advanced fibrosis and cirrhosis, when it has symptoms that overlap with a variety of advanced liver diseases. As such, diagnosis relies entirely on serologies. Given the frequency of HCV in the general population, the asymptomatic nature of early HCV, and the ease of treatment (discussed more below), it is recommended that all adults in the United States be screened for HCV at least once and that high-risk individuals be screened more frequently[ 94 ]. In most patients, diagnostic testing consists of a hepatitis C antibody test with a reflex to HCV RNA viral load if the antibody test is positive. Alternatively, in high-risk patients, some physicians may choose to send an HCV RNA level regardless of antibody result. If any test yields a positive result, further characterization of liver function—including a fibrosis assessment—will help direct further treatment and screening procedures.
The introduction of direct acting antivirals (DAA) has revolutionized the care of patients with HCV[ 95 - 98 ]. With pan-genotypic treatments now available, insurance coverage, falling costs of available agents, and widely available algorithms for simplifying treatment of patients with HCV ( e.g. , https://www.hcvguidelines.org ), treatment of HCV is readily available to the majority of patients in developed countries such as the United States. Treatment goals have therefore shifted towards reaching as many patients as possible and have led to the aggressive goals of eliminating HCV globally[ 25 , 75 ]. As briefly discussed above, an impressive voluntary global health effort in Egypt was able to screen 49.6 million citizens, which identified over 1 million untreated HCV RNA patients and led to successful treatment in 98.8% of patients with long-term follow up[ 79 ]. Remarkably, the cost of identifying and curing each case of Hepatitis C was $130.62 in contrast to the cost of chronic medical care and disability in patients with untreated HCV, which is estimated to be in excess of $100000 per patient[ 79 ]. This study highlights the feasibility of large-scale screening and treatment efforts in resource-limited settings. With the recent changes to the United States Preventative Services Task Force recommendations to expand screening from all adults born between 1945 and 1965 to all adults between age 18 to 79[ 94 ], it will be interesting to see if similar massive-scale screening and treatment can be successfully completed in the United States.
Despite this impressive effort, several factors remain barriers to global elimination of HCV[ 25 ]. Notably, in the Egyptian effort, 20.6% of the population did not participate in voluntary screening with men and young people (< 25 years old) having the lowest participation rates[ 79 ]. While these efforts will likely be successful in patients without ongoing risk factors for HCV infection, IV drug users who are at particularly high risk for HCV infection are among the patients least likely to seek out regular medical care and to adhere to a course of antiviral therapy. With this in mind, multiple studies in the United States have experimented with modified treatment protocols to decrease the burden of treatment and improve access to care. For example, the MINMON study (Clinical Trial Number: NCT03512210 ) aims to test whether a minimal monitoring approach is safe and effective when using the pan-genotypic agent sofosbuvir/velpatasvir in treatment-naïve HCV patients. To do this, they require no pretreatment genotyping, provided patients all of the necessary medication up front, and do not schedule any clinic or lab visits while patients were undergoing treatment but did remotely contact patients at 4 and 22 wk. Promising results for this study were presented at the American Association for the Study of Liver Diseases Liver Meeting in 2020 and showed that sustained virologic response (SVR) was near 95%[ 99 ]. If adopted on a broader scale, this approach has the potential to further simplify HCV treatment and remove some of the treatment burden. Additional studies have attempted to identify the most effective way to treat patients who inject drugs in the multi-center HERO study[ 100 ]. In this study, patients are randomly assigned to receive HCV treatment in one of two ways: (1) Directly observed treatment where patients take medication in front of a staff member; or (2) With the help of patient navigators who attempt to help patients overcome barriers to taking medication. Final results are forthcoming but will hopefully help provide guidance for HCV treatment in this difficult to treat population.
Prevention Efforts
Expansion of HCV prevention strategies are also vital to elimination efforts. Given HCV is most commonly transmitted from unsafe injection practices, especially among injection drug users, programs to increase safe injections are critically important to efforts to prevent HCV transmission and are gaining acceptance[ 101 ].
As with other hepatitis viruses, primary prevention of HCV with vaccination would be extremely beneficial. Moreover, reinfection is a significant risk for patients who have successfully completed DAA therapy but continue to have risk factors for HCV infection. Development of an effective HCV vaccine has proven difficult due to the extreme genetic diversity of HCV—7 known genotypes with over 80 known subtypes—and an error-prone viral polymerase that confers HCV with rapid mutability[ 102 ]. Hope for a vaccine comes from the observation that approximately 25% of acutely infected individuals spontaneously clear HCV[ 103 ]; however, re-infection can occur despite the appearance of broadly neutralizing antibodies in patients who clear their initial HCV infection. Nevertheless, a study that followed 22 active IV drug users who had previously cleared HCV demonstrated that upon reinfection with HCV, virus clearance occurs 83% of the time[ 104 ]. Moreover, reinfection is characterized by reduced maximal viral titers, shorter duration of viremia, and augmented T-cell responses[ 104 ]. Benefit of humoral immunity in preventing HCV infection has been demonstrated in chimpanzees treated with immunoglobulin derived from a human patient in the acute phase of post-transfusion hepatitis C infection[ 105 ]. In this case, human immunoglobulin directed against the hypervariable region 1 of the envelope 2 protein prevented infection with homologous HCV strains[ 105 ]. In another study, HCV neutralizing antibodies derived from a patient infected with HCV genotype 1a protected chimpanzees from infection with genotype 1a and 6a but failed to protect them from infection with HCV genotypes 4a or 5a[ 106 ]. Pools of broadly neutralizing antibodies can prevent infections with multiple HCV genotypes in humanized mice[ 107 , 108 ], and together with the extreme genetic variability of HCV, this suggests that in order for a vaccine to be widely effective, it should be able to induce generation of broadly neutralizing HCV antibodies. Multiple experimental vaccines have used this approach; however, to date, the majority of recipients of these vaccines—either chimpanzee or human—have failed to produce sufficient titers of broadly neutralizing antibodies in most subjects[ 102 , 109 ]. Nevertheless, new culture strategies may enable use of whole inactivated HCV rather than only envelope protein epitopes to allow for additional vaccine epitopes and promote generation of more broadly neutralizing antibodies.
At the moment, prevention of HCV infection is dependent upon behavioral risk reduction ( e.g. , clean needle programs), which is unfortunately being overpowered by a surge of new cases with the ongoing opioid epidemic in the United States. While a vaccine would be ideal, there are various obstacles to overcome, as detailed above. Fortunately, widespread screening and treatment programs are underway on a global scale, and we are hopeful that these will achieve the goal of elimination of chronic hepatitis.
Hepatitis C and Organ Transplantation
DAAs have also made it possible to transplant solid organs from hepatitis C positive donors, which has the potential to greatly expand the donor pool, shorten time on the waitlist, and improve mortality in patients in need of organ transplantation[ 110 ]. Solid organ transplantation can transmit HCV to the organ recipient and was a major concern in the pre-DAA era. This was demonstrated by a retrospective review of all cadaveric donors to the New England Organ Bank between 1986 and 1990, and of 716 organ donors, 1.8% (13) were determined to be HCV positive[ 111 ]. Of these 13 HCV positive donors, their organs—19 kidneys, 6 hearts, and 4 livers—were transplanted into 29 different recipients, and 14 of the recipients (48%) developed HCV, which caused chronic liver disease in the majority of infected patients[ 111 ]. This study raised serious concerns about whether HCV-positive organs should be offered for transplant given the lack of effective treatment for HCV at the time of the study. Subsequent studies from the pre-DAA era showed that transplant patients tend to have worse outcomes if they receive HCV positive organs. HCV-positive liver transplant recipients have decreased patient and graft survival, primarily attributed to HCV recurrence after transplantation[ 112 ]. Additionally, in immunosuppressed patients, including recipients of both liver transplants and other solid organs, HCV positivity is associated with exceptionally high HCV replication and the development of fibrosing cholestatic hepatitis C, an aggressive presentation of HCV that has a high risk of liver failure and mortality[ 113 ]. For heart transplant patients who receive hearts from HCV-positive donors, there is significantly increased mortality at 1, 5, and 10 years that may be attributable to both the development of liver disease and accelerated and more severe coronary artery vasculopathy[ 114 , 115 ]. In kidney transplant recipients, HCV, whether from the donor organ or already present in the recipient, is associated with both increased mortality and graft failure[ 116 , 117 ].
Prior to the introduction of highly effective treatment for HCV, these findings understandably led to reservations about using organs from HCV-positive donors and historically resulted in discarding high-quality organs from HCV-positive donors at much higher rates. Compared to high-quality HCV-negative livers, high-quality HCV-positive livers were 3 times more likely to be discarded from 2005 to 2010, a rate which has decreased after DAAs but is still approximately 1.7 times more likely[ 118 ]. Similarly, high-quality kidneys from HCV-positive donors were 2.6 times more likely to be discarded between 1995 and 2009, and a more recent analysis (from 2005-2014) concluded that only 37% of kidneys from HCV-positive donors were transplanted ( i.e. 63% were discarded) despite most being of good quality and many available recipients[ 119 , 120 ]. Hearts from HCV-positive donors are also significantly less likely to be used. In 2015, hearts from HCV-naïve donors were used approximately 30% of the time compared to 0.7% and 1.4% for hearts from HCV-viremic and non-viremic HCV-Ab positive donors, respectively[ 121 ].
If attitudes towards transplanting solid organs from HCV-positive patients do not change, there is likely to be an even greater mismatch between available high-quality donor organs and patients on transplant waitlists, as the opioid epidemic is resulting in an alarming rise in overdose deaths[ 122 ]. A recent National Registry Study characterized organ donations form deceased donors who died of drug overdose (ODD, overdose death donor) between 2000 and 2017 found that ODDs have risen significantly in that time from 1.1% of donors in 2000 to 13.4% of donors in 2017[ 122 ]. Compared to non-ODDs, ODDs were more likely to be young, white, have little comorbidity, and have HCV infection (18.3% were HCV-positive)[ 122 ]. Despite the higher rates of HCV positivity, the authors found similar 5-year patient and graft survival rates between non-ODDs and ODDs[ 122 ].
Along with the organ shortage and rise in availability of high-quality organs from HCV-positive donors, the success of DAAs in curing HCV has led to several clinical trials to determine the outcomes of transplanting organs from HCV-positive donors. In general, these trials have been an overwhelming success. Two separate studies of 10 HCV-mismatched kidney transplant recipients (Donor positive, recipient negative) showed 100% success rate of achieving SVR in recipients[ 123 , 124 ]. Moreover, this mismatched strategy reduced wait times, and recipients had excellent graft function at 6 mo post-transplant[ 124 ]. Similar results have been shown in both mismatched heart and lung transplant patients. An early study performed HCV-mismatched heart transplants on 13 patients, and 9 of the 13 developed HCV viremia shortly after transplant. Eight of these patients completed DAA treatment and demonstrated SVR (the 9 th patient died of a pulmonary embolus)[ 125 ]. Another study enrolled 44 patients awaiting heart or lung transplant and performed 36 HCV-mismatched lung transplants and 9 heart transplants regardless of HCV genotype[ 126 ]. Ninety-five percent of patients had detectable HCV RNA immediately after transplant, and 100% achieved SVR with the pan-genotypic DAA sofosbuvir–velpatasvir[ 126 ]. Importantly, at 6 mo, the patients continued to have excellent graft function and no detectable HCV RNA[ 126 ].
Finally, a larger, more recent study that performed 80 HCV-mismatched heart transplants and followed patients for 1 year showed comparable 1-year survival and similarly, low rates of allograft rejection between patients who received HCV-positive organs and those who received HCV-negative organs[ 127 ]. However, there was significantly more primary graft dysfunction and a trend towards increased early coronary allograft vasculopathy in patients who received organs from HCV-positive donors, although this was not statistically significant[ 127 ]. A large retrospective registry study of patients who underwent a single organ liver transplant from 2008 through 218 demonstrated similar 2-year graft survival rates (approximately 88%) among all combinations of donor-recipient HCV RNA status[ 128 ]. Importantly, graft survival from HCV-positive donors has improved in the DAA era, with 3-year graft survival now approximately 88% regardless of recipient HCV status[ 128 ]. Moreover, this study demonstrated a trend towards increasing use of organs from HCV-positive donors for HCV-negative recipients from 7 in 2008 to 107 in 2018, highlighting increased acceptance of this novel strategy of expanding the pool of available organs[ 128 ].
Together, these data are extremely promising in demonstrating early, post-transplant HCV cure and similar graft and recipient survival in short- and mid-term follow-up. They also provide justification for the use of organs from HCV-positive donors, regardless of recipient HCV status. This strategy is gaining popularity: The Scientific Registry Transplant Recipients indicates increasing numbers of mismatched transplants, and there are now similar utilization rates of available HCV-positive and HCV-negative donor hearts[ 128 ]. Nevertheless, there still exists some skepticism of this strategy, as a recent survey of 99 transplant nephrology providers demonstrated that fewer than half support offering HCV-positive kidneys as part of routine care outside of a research setting[ 129 ]. These attitudes will likely shift to usher in more widespread use of this practice if long-term outcomes are as promising as the available clinical trial results.
HEPATITIS D
Hepatitis D infection is caused by the Hepatitis D virus (HDV), a single stranded, enveloped RNA molecule. HDV is the smallest virus known to infect humans, and it is often classified as a subvirus given that the HDV lifecycle is entirely dependent on HBV[ 130 , 131 ]. Transmission occurs through similar means as HBV and can occur either at the same time as an HBV infection ( i.e. , coinfection) or in patients with chronic HBV infections ( i.e. , superinfection). This relationship to HBV infection timing determines the natural history of HDV infection, with superinfection more often leading to a rapid clinical deterioration with progressive hepatitis, cirrhosis and development of complications of cirrhosis, including HCC. Current treatment strategies for HDV are centered around prevention and the management of HBV, as HDV is entirely dependent on the HBV lifecycle; however, directed therapies against HDV are under investigation[ 132 ].
There are multiple estimates of HDV prevalence, as high as 13% of all HBV carriers; however, a recent meta-analysis estimated that approximately 4.5% of HBsAg-positive people are coinfected with HDV, which corresponds to approximately 12 million HDV infections worldwide[ 133 ]. HDV is present worldwide with geographic variation that does not align with HBV prevalence. There is a very high prevalence of HDV in Mongolia (36.9% of patients with HBsAg) and in central African countries (with estimates of > 10% prevalence in the HBsAg-positive population). Despite high rates of HBV infection, there are very low rates of HDV co-infection in other Asian countries, including China[ 133 ]. Regardless of geography, the populations at highest risk for HDV include people who inject drugs and those with human immunodeficiency virus (HIV) or HCV[ 133 ]. HDV is even more prevalent in patients with HBV-associated cirrhosis and HCC, highlighting the pathogenic importance of HDV.
The primary modes of transmission for HDV are similar to those for HBV with the highest infection rates in people who inject drugs. HDV is a 1.7kb single stranded RNA molecule that encodes for the hepatitis D antigen (HDAg)[ 134 ]. Depending on RNA processing, the HDAg has one of two forms—the short form (HDAg-S) or long form (HDAg-L)—each with distinct functional roles. HDAg-S activates further HDV RNA synthesis, while HDAg-L inhibits HDV RNA synthesis and promotes HDV assembly with HBsAg to allow for packaging and transport[ 134 ]. It has been proposed that HDV mediates liver toxicity via both direct hepatotoxicity and indirect immune-mediated hepatocellular damage, although immune-mediated damage is the current prevailing theory.
Evidence suggesting direct hepatotoxicity is based on (1) Histopathology with limited immune infiltrate in acute HDV infection[ 135 ]; (2) ultrastructural analysis of hepatocytes infected with HBV and with HDV that showed a strong correlation between the appearance of cytoplasmic structures and hepatocellular damage as assessed by ALT levels[ 136 ]; and (3) in vitro cell culture data in which HDAg expression is associated with cell death in the absence of immune cells[ 137 ]. However, the conclusion that HDV causes direct cytotoxicity has been questioned. Transgenic mice with hepatocyte-specific expression of both HDAg-S and HDAg-L demonstrated no direct cytotoxic effect regardless of the level of HBsAg coexpression[ 138 ]. This finding was mirrored with histologic assessment of HBsAg carriers with chronic HDV infection, which demonstrated that HDAg expression was low during acute hepatitis and increased with the development of chronic disease[ 139 ]. However, these studies do not test the possibility of other stages of the HDV lifecycle causing direct cytotoxicity and some have suggested that HDV viral replication itself leads to cytotoxicity. More convincing evidence is present for HDV infection causing immune-mediated hepatotoxicity. Nevertheless, long-term follow up of 76 patients who underwent liver transplant for HDV-related cirrhosis showed that either HDV RNA (serum) or liver HDAg were present in 88% of patients within the first year[ 140 ]. However, these patients did not develop hepatitis unless active HBV infection recurs, suggesting that HDV is not hepatotoxic when expressed alone but instead requires HBV expression[ 140 ].
Regardless of the mode of hepatotoxicity, HDV has different clinical courses that depend on the timing of HDV infection in relationship to HBV-mediated liver disease. For coinfection, HBV and HDV are acquired simultaneously, whereas a superinfection occurs when HDV is acquired in a patient with an established HBV infection. Coinfection is most common in patients who use IV drugs. Since HDV is dependent on HBV and HBV infection is cleared in the majority of cases acquired in adulthood, HDV acquired in this manner also spontaneously clears. When the coinfection does not clear, some evidence suggests that HDV may cause a more intense hepatitis[ 141 ]. This was suggested by the high seroprevalence of HDV in patients with fulminant hepatitis B (52% in 1 study), but in this same study of fulminant hepatitis B, HDV coinfection correlated with better survival than with those infected with HBV alone (57.8% survival in coinfection vs. 16.7% survival with HBV alone)[ 141 ].
HDV superinfection occurs when a patient with a preexisting liver disease from chronic HBV infection contracts HDV. This is often accompanied by very high levels of HDV RNA expression with a subsequent severe hepatitis and decompensation of preexisting liver disease, as the HDV replication is protected by high levels of HBsAg[ 142 ]. Despite HDV inhibition of HBV replication, HBV infection is already established and allows development of chronic HDV infection in the majority of cases. Chronic HDV infection often leads to hepatitis with rapidly developing cirrhosis and increased risk of decompensation and HCC[ 143 ]. The increased risk of HCC in patients chronically infected with HDV is evidenced by multiple long-term cohort studies. A 28-year study of 299 patients with HDV infection with a median follow up of 233 mo demonstrated that active HDV replication portends an increased risk of cirrhosis and HCC with annual rates of 4% and 2.8%, respectively[ 144 ]. Moreover, this study also reported that HDV replication was an independent predictor of mortality in these patients[ 144 ]. Another cohort demonstrated that HCC developed in 42% of patients with chronic HDV within 12 years of diagnosis[ 131 ].
As discussed above, HDV infection and replication is entirely dependent upon HBV infection, and therefore HDV prevention efforts are largely centered around HBV prevention with vaccination programs. HBV vaccination programs have therefore led to a significant reduction in HDV infections in developed countries[ 145 ]. The impressive ongoing global efforts to eliminate chronic viral hepatitis—including HBV—should also significantly reduce the burden of HDV.
The only currently available treatment for HDV is interferon, which may exert its effect on HDV either via direct inhibition of HDV or inhibition of HBV[ 146 ]. Despite initial control of HDV viral load and improved histology during the treatment phases, multiple trials have shown exceptionally high rates of HDV relapse after completing interferon therapy[ 147 ]. The largest of these studies randomly assigned 42 patients with chronic HDV to receive either placebo or 3 million or 9 million units of interferon-alpha-2a[ 147 ]. This study demonstrated a dose-dependent response with regards to ALT levels, HDV RNA levels, and histology scores[ 147 ]. While the decreases in ALT levels were maintained for 4 years of follow up, even patients treated with high dose interferon demonstrated relapses in HDV replication during post-treatment follow up[ 147 ]. Nevertheless, when these patients were followed for an additional 2-14 years, this group discovered that patients who received high dose interferon therapy had a dramatic long-term survival benefit[ 148 ]. Further, non-randomized, studies assessed the response of chronic HDV to peginterferon-alpha-2b[ 149 , 150 ]. These studies demonstrated variable responses to peginterferon-alpha-2b with a 57% sustained response in one study and only 17% sustained response in the other study with sustained response defined as undetectable HDV-RNA 6-mo post-treatment[ 149 , 150 ]. Those most likely to have a sustained response are likely to also have early reductions in HDV-RNA levels[ 149 , 150 ]. Conversely, no patients with cirrhosis demonstrated a sustained response[ 149 ].
Given the dependence of HDV on HBV, it is logical that therapies directed against HBV may also treat HDV. However, HDV replication is independent of HBV replication and only requires HBsAg. Therefore, any therapeutic benefit of HBV therapy on HDV infection is an indirect result of decreasing HBsAg production, which is not significantly reduced by nucleos(t)ide analogs. Unfortunately, trials of nucleos(t)ide analogs have not proven any sustained benefit in chronic hepatitis D despite excellent suppression of HBV DNA[ 151 ]. While there have been reports of Tenofovir resulting in HDV virus suppression in patients with either HBV-HDV co-infection or triple infection with HIV, HBV, and HDV[ 152 , 153 ], but it is questioned whether this results in a sustained benefit if tenofovir is withdrawn. Moreover, when combined with peginterferon alpha-2a in a placebo-controlled randomized control trial, tenofovir had no additional benefit with respect to HDV RNA level[ 154 ]. Similarly, the nucleoside analog adefovir also had no benefit on HDV levels when used alone and had no added benefit to interferon therapy[ 155 ]. Taken together, these data highlight the need for more directed HDV therapies. Directed treatments against HDV are currently under investigation and included inhibitors of cell entry, prenylation inhibitors, and subviral particle release inhibitors[ 132 , 146 ]. However, none of these are currently approved for use outside of clinical trials.
HEPATITIS E
Hepatitis E infection is caused by the Hepatitis E virus (HEV), a single stranded, quasi-enveloped RNA molecule in the Hepeviridae family[ 156 ]. There are 8 known genotypes, with genotypes 1-4 being the most studied[ 157 ]. Transmission occurs either via fecal-oral route (genotypes 1 and 2) or zoonotic transmission with ingestion of raw or undercooked meat (genotypes 3 and 4). HEV typically causes an acute, self-limited hepatitis that is particularly severe in pregnant women; however, cases of chronic hepatitis E have been reported in immunocompromised hosts. Prevention is centered around improving hygiene and vaccination, although a vaccine is available for use only in China[ 158 ]. Currently, treatment of acute HEV is supportive, and chronic HEV is treated with a multipronged approach with decreasing the underlying immunosuppression and occasionally ribavirin.
There are two distinct, genotype-dependent epidemiologic patterns of Hepatitis E. Hepatitis E virus genotypes 1 and 2 are only known to infect humans and are highly endemic in developing countries throughout the world[ 157 ]. In these regions, HEV genotypes 1 and 2 result in large outbreaks in areas of poor sanitation, primarily in developing countries and related to drinking water contaminated with feces[ 159 ]. This was demonstrated by a naïve volunteer who developed a hepatitis-like illness after ingesting pooled feces extracts from patients with non-A, non-B hepatitis, which led to the discovery of hepatitis E[ 160 ]. Genotypes 1 and 2 are estimated to cause at least 20 million acute infections that result in approximately 3.4 million symptomatic cases, 70000 deaths, and 3000 stillbirths each year[ 161 ].
In addition to humans, HEV genotypes 3 and 4 are known to infect multiple species including swine, deer, and rabbits and are more commonly reported in the developed world (Europe, North America, Australia, and Japan)[ 156 ]. HEV genotypes 3 and 4 account for very few (less than 1%) of the cases of viral hepatitis in the developed world, and transmission is thought to be from consumption of undercooked meat, primarily pork. This is evidenced by the exceptionally high rate of HEV infection in both wild and domesticated pigs in multiple European countries[ 162 , 163 ]. A study of the Swedish swine population also sequenced partial genomes of HEV strains from humans, pigs, and wild boars and found a high degree of relatedness, suggesting zoonotic transmission[ 163 ]. The ability of swine HEV genotype 3 to cross species was confirmed with inoculation of rhesus monkeys and chimpanzees[ 164 ]; however, pigs have varying degrees of susceptibility to experimental infection with HEV from humans[ 165 , 166 ]. Nevertheless, HEV has been detected in multiple stages of consumer pork product processing across Europe[ 167 - 169 ], but HEV is inactivated by adequate food preparation, as pigs cannot be inoculated with HEV-infected pureed pig livers subjected to heating to 71 °C for 20 min or longer[ 170 ].
While very few cases of HEV are reported in the developed world each year, this likely owes to undertesting and the self-limited nature of the disease. Screening blood donations in Western Europe has shown HEV RNA in 0.02%-0.14% of all screened blood products. In England, one study that screened 225000 blood donations found that 79 donors (0.04%) were infected with HEV RNA[ 171 ]. These infected donors generated in 129 blood components and resulted in an estimated 42% infectivity rate (18 of 43 followed up), which has led some to call for universal screening of blood donations for HEV[ 171 , 172 ]. Seropositivity in the developed world ranges from 6% in the United States to 39.1% in Southern France, although assays for HEV IgG have reportedly wide ranges of sensitivity[ 156 ]. A 2017 systematic review that analyzed the prevalence of HEV in Iran found an overall positivity rate of 9.7% with varying rates between cities (1.1% in Tehran vs 46.1% in Ahvaz)[ 173 ]. Finally, a recent systematic review and meta-analysis of 56 studies on the seroprevalence of HEV in patients on maintenance hemodialysis (HD) found that the pooled seroprevalence of HEV was 11.13% between 2016 and 2020, which had increased from 6.6% from 1994 to 2000[ 174 ]. Moreover, the length of time on HD was associated with higher seroprevalence as those on HD for more than 60 mo had a significantly higher chance of being seropositive (27.69% more than 60 mo vs 15.78% less than 60 mo)[ 174 ].
Hepatitis E enters into the hepatocyte in 1 of 2 distinct mechanisms depending on whether the virus is enveloped or not, and upon entry, the virus is uncoated, transcribed and translated into 3 different proteins that facilitate viral replication, repackaging, sorting and release into both the blood stream and bile[ 156 ]. Upon HEV release into the bile, the envelope is degraded by bile acids and detergents prior to being released into the intestinal lumen and excreted with stool. The unenveloped version of the virus released into the feces has a much more efficient mechanism of hepatocyte entry and helps explain the most common form of HEV transmission. HEV is not thought to be directly cytotoxic. Instead, HEV causes hepatotoxicity via immune mediated mechanisms as patients with acute hepatitis E infection show both CD4 + and CD8 + T-cell responses[ 175 ].
HEV infection is most commonly asymptomatic as evidenced by the high seroprevalence of anti-HEV IgG and lack of recollection of acute symptoms[ 176 ]. When symptomatic, HEV has a 2-10 d incubation period followed by non-specific prodromal symptoms of fatigue, nausea, vomiting, and anorexia, similar to other acute hepatitis infections. Ultimately, HEV causes an acute, self-limited hepatitis characterized by jaundice and marked AST and ALT elevations. These symptoms typically self-resolve within 1 mo, although some cases of prolonged cholestasis (up to 6 mo) and even cases of fulminant hepatic failure with resultant death have been reported[ 177 ].
Pregnant women are particularly prone to severe hepatitis E infections that can result in fulminant hepatic failure in approximately 20% of infected patients with an associated high mortality rate and high rate of fetus and neonatal complications, including spontaneous abortions, stillbirths, and neonatal deaths. Infections within the third trimester are the most problematic and are thought to be so detrimental because of an altered immune system and hormonal changes[ 178 ].
All reported cases of chronic hepatitis E infection have been linked to genotype 3 and have been reported in immunocompromised patients including those with HIV[ 179 ], undergoing chemotherapy[ 180 ], or solid organ transplant recipients[ 181 ]. This has been linked to the development of cirrhosis and HCC. However, the limited number of cases reported precludes detailed assessment of risk of these complications of chronic HEV infection.
Given that the primary mode of transmission for the most common HEV variants (genotypes 1 and 2) is through contaminated water, the main mode of prevention is through increased sanitation efforts to provide citizens of the developing world with clean drinking water. A vaccine to protect against HEV is now approved for human use in China but not elsewhere; however, this is likely to benefit those infected in the rest of the world[ 158 ]. Efforts to bring clean drinking water to all parts of the world should reduce the burden of HEV, and we are hopeful that the vaccine for HEV may be approved outside of China for further HEV prevention.
There is currently no direct acting antiviral to treat HEV. In immunocompromised patients, the mainstay of treatment is to reduce immunosuppression if possible[ 181 , 182 ]. This results in a sustained viral response ( i.e. , no detectable HEV RNA) in approximately 30% of patients. In patients where decreasing immunosuppression is either not possible or does not result in SVR, ribavirin is currently the main therapeutic option[ 183 ]. Unfortunately, ribavirin is a category X drug, meaning that is should not be given to pregnant women. Some cases of ribavirin exposure do occur during pregnancy, which is the subject of study in the ribavirin pregnancy registry[ 184 ]. Interim analysis has failed to show a direct link between ribavirin and teratogenicity, but full results of the study are forthcoming and for the moment, ribavirin remains a category X drug[ 184 ]. Additional therapies, including pegylated interferon and sofosbuvir have been evaluated only in limited clinical settings[ 185 , 186 ].
HEPATITIS G
What is now regarded as the hepatitis G virus (HGV) was initially described in 1966 and named the GB virus (GBV) after the surgeon G. Barker, who fell ill with hepatitis. Subsequent studies isolated genetically similar agents, named GBV-A, GBV-B, and GBV-C, and GBV-C was genetically similar to a separate isolate already named HGV[ 187 ].
Hepatitis G virus is a less well-described entity with a global distribution but varying reports of HGV prevalence in the general population. A systematic review evaluated studies reporting the prevalence of HGV in healthy voluntary blood donors and found 649 infections in 13610 voluntary blood donors (4.8% positivity rate) [ 188 ]. The studies included in the review reported HGV positivity rates ranging from 0.5% prevalence in a Japanese study to 18.9% in a South African study[ 188 ]. Currently, HGV is not widely tested for clinically, so the true incidence is not well-known.
The natural history of HGV is not well-characterized, and it is questioned whether HGV causes a clinically significant liver disease. While HGV has been detected in many different clinical scenarios, including acute presumed viral hepatitis, cirrhosis of unknown cause, and HCC, the clinical relevance is still questioned given the high co-infection rate with other causes of viral hepatitis, including HCV and that HGV co-infection does not exacerbate the course of HCV[ 189 ]. Moreover, greater than 75% of patients who are positive for HGV have normal liver function tests[ 190 ]. This group also reported that among HGV-positive individuals with elevated liver function tests, aminotransferase elevations are negligible and that there is no correlation between HGV viral load and aminotransferase elevations in patients on chronic dialysis[ 190 ]. In another study, 0 of 16 HGV-positive patients on chronic hemodialysis had elevated aminotransferase levels despite prolonged viral persistence[ 191 ].
Additionally, individuals who undergo liver transplantation are often incidentally HGV-positive, and both HGV-positive and HGV-negative liver recipients have similar outcomes (including 40-mo survival), suggesting that HGV positivity does not affect engraftment or graft function[ 192 ]. Additional studies have suggested that the liver is not the site of viral replication as the liver-to-serum ratio of HGV RNA is less than 1, which is consistent with blood contamination of liver tissue[ 193 ]. Finally, despite initial reports of HGV-associated fulminant liver failure, it is now thought that these patients were positive for HGV RNA due to blood product administration during the hospitalization rather than HGV being present at the time of admission[ 194 ]. Experimental models also argue against HGV causing a clinically significant disease as studies in chimpanzees demonstrate that HGV can reach high levels of viremia in chimpanzees without causing a concurrent rise in liver enzymes[ 195 ]. Taken together, these data provide evidence that HGV does not cause clinically significant liver disease. As such, HGV is not a common test ordered on the clinical wards.
As a group, viral hepatitis represents an ongoing global health concern (Table 1 ). Acute viral hepatitis infections—HAV and HEV—tend to be self-limited infections with little-to-no long-lasting effect, and both vaccines and improved sanitation conditions will decrease the burden of disease over time. Moreover, ever-improving understanding of the risk factors for acute liver failure from acute hepatitis and experimental supportive care options will aid in further reducing the impact of acute viral hepatitis. Chronic viral hepatitis infections—HBV and HCV—on the other hand, often result in cirrhosis and death if left untreated. While vaccination for HBV is highly effective in reducing transmission, and treatments for HBV are effective in reducing HBV viral load and progression of liver disease, an effective cure for HBV remains elusive. Conversely, an effective vaccine for HCV has not been achieved, but highly effective treatments cure HCV in nearly 100% of cases and are now revolutionizing the fields of transplant medicine. Despite unique barriers, there are ongoing global efforts to eliminate chronic viral hepatitis, and given substantial progress, we are hopeful that this ambitious goal will be realized.
Overview of epidemiology, symptoms, natural history and clinical management of viral hepatitis infections
HAV: Hepatitis A virus; HBV: Hepatitis B virus; HCV: Hepatitis C virus; HDV: Hepatitis D virus; HEV: Hepatitis E virus; HGV: Hepatitis G virus; WHO: World Health Organization; IVDU: Intravenous drug use; HCC: Hepatocellular carcinoma; HBIG Hepatitis B immunoglobulin; HBsAb: Hepatitis B surface antibody; HBsAg: Hepatitis B surface antigen; IgG: Immunoglobulin G; IgM: Immunoglobulin M.
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: June 27, 2021
First decision: July 14, 2021
Article in press: March 4, 2022
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: United States
Peer-review report’s scientific quality classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Cao HC, China; Wang CR, Taiwan S-Editor: Chang KL L-Editor: A P-Editor: Chang KL
Contributor Information
Matthew August Odenwald, Department of Medicine, Section of Gastroenterology, Hepatology, and Nutrition, Center for Liver Diseases, University of Chicago, Chicago, IL 60637, United States.
Sonali Paul, Department of Medicine, Section of Gastroenterology, Hepatology, and Nutrition, Center for Liver Diseases, University of Chicago, Chicago, IL 60637, United States. [email protected].
- 1. Coulepis AG, Locarnini SA, Westaway EG, Tannock GA, Gust ID. Biophysical and biochemical characterization of hepatitis A virus. Intervirology. 1982;18:107–127. doi: 10.1159/000149314. [ DOI ] [ PubMed ] [ Google Scholar ]
- 2. Coulepis AG, Anderson BN, Gust ID. Hepatitis A. Adv Virus Res. 1987;32:129–169. doi: 10.1016/s0065-3527(08)60476-5. [ DOI ] [ PubMed ] [ Google Scholar ]
- 3. Jacobsen KH, Wiersma ST. Hepatitis A virus seroprevalence by age and world region, 1990 and 2005. Vaccine. 2010;28:6653–6657. doi: 10.1016/j.vaccine.2010.08.037. [ DOI ] [ PubMed ] [ Google Scholar ]
- 4. Rezende G, Roque-Afonso AM, Samuel D, Gigou M, Nicand E, Ferre V, Dussaix E, Bismuth H, Féray C. Viral and clinical factors associated with the fulminant course of hepatitis A infection. Hepatology. 2003;38:613–618. doi: 10.1053/jhep.2003.50366. [ DOI ] [ PubMed ] [ Google Scholar ]
- 5. Cuthbert JA. Hepatitis A: old and new. Clin Microbiol Rev. 2001;14:38–58. doi: 10.1128/CMR.14.1.38-58.2001. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 6. The World Health Organization. Immunization, Vaccines and Biologicals: Hepatitis A. Available from: https://www.who.int/immunization/diseases/hepatitisA/en/
- 7. Klevens RM, Kruszon-Moran D, Wasley A, Gallagher K, McQuillan GM, Kuhnert W, Teshale EH, Drobeniuc J, Bell BP. Seroprevalence of hepatitis A virus antibodies in the U.S.: results from the National Health and Nutrition Examination Survey. Public Health Rep. 2011;126:522–532. doi: 10.1177/003335491112600408. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 8. Bell BP, Kruszon-Moran D, Shapiro CN, Lambert SB, McQuillan GM, Margolis HS. Hepatitis A virus infection in the United States: serologic results from the Third National Health and Nutrition Examination Survey. Vaccine. 2005;23:5798–5806. doi: 10.1016/j.vaccine.2005.03.060. [ DOI ] [ PubMed ] [ Google Scholar ]
- 9. Centers for Disease Control and Prevention. Hepatitis A. Available from: https://www.cdc.gov/vaccines/pubs/pinkbook/hepa.html .
- 10. Rosenblum LS, Villarino ME, Nainan OV, Melish ME, Hadler SC, Pinsky PP, Jarvis WR, Ott CE, Margolis HS. Hepatitis A outbreak in a neonatal intensive care unit: risk factors for transmission and evidence of prolonged viral excretion among preterm infants. J Infect Dis. 1991;164:476–482. doi: 10.1093/infdis/164.3.476. [ DOI ] [ PubMed ] [ Google Scholar ]
- 11. Provost PJ, Hilleman MR. Propagation of human hepatitis A virus in cell culture in vitro. Proc Soc Exp Biol Med. 1979;160:213–221. doi: 10.3181/00379727-160-40422. [ DOI ] [ PubMed ] [ Google Scholar ]
- 12. Phan C, Hollinger FB. Hepatitis A: Natural history, immunopathogenesis, and outcome. Clin Liver Dis (Hoboken) 2013;2:231–234. doi: 10.1002/cld.253. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 13. Taylor RM, Davern T, Munoz S, Han SH, McGuire B, Larson AM, Hynan L, Lee WM, Fontana RJ US Acute Liver Failure Study Group. Fulminant hepatitis A virus infection in the United States: Incidence, prognosis, and outcomes. Hepatology. 2006;44:1589–1597. doi: 10.1002/hep.21439. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 14. Kim JD, Cho EJ, Ahn C, Park SK, Choi JY, Lee HC, Kim DY, Choi MS, Wang HJ, Kim IH, Yeon JE, Seo YS, Tak WY, Kim MY, Lee HJ, Kim YS, Jun DW, Sohn JH, Kwon SY, Park SH, Heo J, Jeong SH, Lee JH, Nakayama N, Mochida S, Ido A, Tsubouchi H, Takikawa H, Shalimar , Acharya SK, Bernal W, O'Grady J, Kim YJ. A Model to Predict 1-Month Risk of Transplant or Death in Hepatitis A-Related Acute Liver Failure. Hepatology. 2019;70:621–629. doi: 10.1002/hep.30262. [ DOI ] [ PubMed ] [ Google Scholar ]
- 15. Rela M, Kaliamoorthy I, Reddy MS. Current status of auxiliary partial orthotopic liver transplantation for acute liver failure. Liver Transpl. 2016;22:1265–1274. doi: 10.1002/lt.24509. [ DOI ] [ PubMed ] [ Google Scholar ]
- 16. WHO position paper on hepatitis A vaccines - June 2012. Wkly Epidemiol Rec. 2012;87:261–76. [ PubMed ] [ Google Scholar ]
- 17. Desai AN, Kim AY. Management of Hepatitis A in 2020-2021. JAMA. 2020;324:383–384. doi: 10.1001/jama.2020.4017. [ DOI ] [ PubMed ] [ Google Scholar ]
- 18. Victor JC, Monto AS, Surdina TY, Suleimenova SZ, Vaughan G, Nainan OV, Favorov MO, Margolis HS, Bell BP. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med. 2007;357:1685–1694. doi: 10.1056/NEJMoa070546. [ DOI ] [ PubMed ] [ Google Scholar ]
- 19. Nelson NP, Link-Gelles R, Hofmeister MG, Romero JR, Moore KL, Ward JW, Schillie SF. Update: Recommendations of the Advisory Committee on Immunization Practices for Use of Hepatitis A Vaccine for Postexposure Prophylaxis and for Preexposure Prophylaxis for International Travel. MMWR Morb Mortal Wkly Rep. 2018;67:1216–1220. doi: 10.15585/mmwr.mm6743a5. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 20. Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [ DOI ] [ PubMed ] [ Google Scholar ]
- 21. Kruszon-Moran D, Paulose-Ram R, Martin C, Barker L, and McQuillan, G Prevalence and trends in hepatitis B virus infection in the United States, 2015-2018. NCHS Data Brief. 2020;361:1–8. [ PubMed ] [ Google Scholar ]
- 22. Jefferies M, Rauff B, Rashid H, Lam T, Rafiq S. Update on global epidemiology of viral hepatitis and preventive strategies. World J Clin Cases. 2018;6:589–599. doi: 10.12998/wjcc.v6.i13.589. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 23. McMahon BJ, Alward WL, Hall DB, Heyward WL, Bender TR, Francis DP, Maynard JE. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis. 1985;151:599–603. doi: 10.1093/infdis/151.4.599. [ DOI ] [ PubMed ] [ Google Scholar ]
- 24. Fanning GC, Zoulim F, Hou J, Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov. 2019;18:827–844. doi: 10.1038/s41573-019-0037-0. [ DOI ] [ PubMed ] [ Google Scholar ]
- 25. Thomas DL. Global Elimination of Chronic Hepatitis. N Engl J Med. 2019;380:2041–2050. doi: 10.1056/NEJMra1810477. [ DOI ] [ PubMed ] [ Google Scholar ]
- 26. Shepard CW, Simard EP, Finelli L, Fiore AE, Bell BP. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol Rev. 2006;28:112–125. doi: 10.1093/epirev/mxj009. [ DOI ] [ PubMed ] [ Google Scholar ]
- 27. Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet. 2015;386:1546–1555. doi: 10.1016/S0140-6736(15)61412-X. [ DOI ] [ PubMed ] [ Google Scholar ]
- 28. Mitchell T, Armstrong GL, Hu DJ, Wasley A, Painter JA. The increasing burden of imported chronic hepatitis B--United States, 1974-2008. PLoS One. 2011;6:e27717. doi: 10.1371/journal.pone.0027717. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 29. Tavakoli A, Moghoofei M, Mostafaei S, Ghaffari H, Alavian SHM& SM, Alavian SM. Prevalence of hepatitis B surface antigen among hemodialysis patients from Middle Eastern countries: a systematic review and meta-analysis. Future Virology. 2017:12. [ Google Scholar ]
- 30. Oh IS, Park SH. Immune-mediated Liver Injury in Hepatitis B Virus Infection. Immune Netw. 2015;15:191–198. doi: 10.4110/in.2015.15.4.191. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 31. Tan A, Koh S, Bertoletti A. Immune Response in Hepatitis B Virus Infection. Cold Spring Harb Perspect Med. 2015;5:a021428. doi: 10.1101/cshperspect.a021428. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 32. Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A. 2004;101:6669–6674. doi: 10.1073/pnas.0401771101. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 33. Tan G, Song H, Xu F, Cheng G. When Hepatitis B Virus Meets Interferons. Front Microbiol. 2018;9:1611. doi: 10.3389/fmicb.2018.01611. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 34. Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, Brown D, Gilson RJ, Tedder RJ, Dusheiko GM, Jacobs M, Klenerman P, Maini MK. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology. 2009;137:1289–1300. doi: 10.1053/j.gastro.2009.06.054. [ DOI ] [ PubMed ] [ Google Scholar ]
- 35. Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 36. Maini MK, Boni C, Ogg GS, King AS, Reignat S, Lee CK, Larrubia JR, Webster GJ, McMichael AJ, Ferrari C, Williams R, Vergani D, Bertoletti A. Direct ex vivo analysis of hepatitis B virus-specific CD8(+) T cells associated with the control of infection. Gastroenterology. 1999;117:1386–1396. doi: 10.1016/s0016-5085(99)70289-1. [ DOI ] [ PubMed ] [ Google Scholar ]
- 37. Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 38. Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, Swadling L, Alberts E, Davidson BR, Kennedy PT, Gill US, Mauri C, Blair PA, Pelletier N, Maini MK. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest. 2018;128:4588–4603. doi: 10.1172/JCI121960. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 39. Heim K, Neumann-Haefelin C, Thimme R, Hofmann M. Heterogeneity of HBV-Specific CD8+ T-Cell Failure: Implications for Immunotherapy. Front Immunol. 2019;10:2240. doi: 10.3389/fimmu.2019.02240. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 40. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, Zerbini A, Cavalli A, Missale G, Bertoletti A, Ferrari C. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 41. de Jongh FE, Janssen HL, de Man RA, Hop WC, Schalm SW, van Blankenstein M. Survival and prognostic indicators in hepatitis B surface antigen-positive cirrhosis of the liver. Gastroenterology. 1992;103:1630–1635. doi: 10.1016/0016-5085(92)91188-a. [ DOI ] [ PubMed ] [ Google Scholar ]
- 42. Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, Brown RS Jr, Bzowej NH, Wong JB. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67:1560–1599. doi: 10.1002/hep.29800. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 43. Lok AS, Zoulim F, Dusheiko G, Chan HLY, Buti M, Ghany MG, Gaggar A, Yang JC, Wu G, Flaherty JF, Subramanian GM, Locarnini S, Marcellin P. Durability of Hepatitis B Surface Antigen Loss With Nucleotide Analogue and Peginterferon Therapy in Patients With Chronic Hepatitis B. Hepatol Commun. 2020;4:8–20. doi: 10.1002/hep4.1436. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 44. Perrillo RP, Brunt EM. Hepatic histologic and immunohistochemical changes in chronic hepatitis B after prolonged clearance of hepatitis B e antigen and hepatitis B surface antigen. Ann Intern Med. 1991;115:113–115. doi: 10.7326/0003-4819-115-2-113. [ DOI ] [ PubMed ] [ Google Scholar ]
- 45. Loomba R, Liang TJ. Hepatitis B Reactivation Associated With Immune Suppressive and Biological Modifier Therapies: Current Concepts, Management Strategies, and Future Directions. Gastroenterology. 2017;152:1297–1309. doi: 10.1053/j.gastro.2017.02.009. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 46. The World Health Organization. Global Hepatitis Report, 2017. [cited 27 June 2021]. Available from: https://www.who.int/publications/i/item/global-hepatitis-report-2017 .
- 47. Chen HL, Chang MH, Ni YH, Hsu HY, Lee PI, Lee CY, Chen DS. Seroepidemiology of hepatitis B virus infection in children: Ten years of mass vaccination in Taiwan. JAMA. 1996;276:906–908. [ PubMed ] [ Google Scholar ]
- 48. Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, Kong MS, Liang DC, Shau WY, Chen DS. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855–1859. doi: 10.1056/NEJM199706263362602. [ DOI ] [ PubMed ] [ Google Scholar ]
- 49. McMahon BJ, Bulkow LR, Singleton RJ, Williams J, Snowball M, Homan C, Parkinson AJ. Elimination of hepatocellular carcinoma and acute hepatitis B in children 25 years after a hepatitis B newborn and catch-up immunization program. Hepatology. 2011;54:801–807. doi: 10.1002/hep.24442. [ DOI ] [ PubMed ] [ Google Scholar ]
- 50. Beasley RP, Hwang LY, Lee GC, Lan CC, Roan CH, Huang FY, Chen CL. Prevention of perinatally transmitted hepatitis B virus infections with hepatitis B immune globulin and hepatitis B vaccine. Lancet. 1983;2:1099–1102. doi: 10.1016/s0140-6736(83)90624-4. [ DOI ] [ PubMed ] [ Google Scholar ]
- 51. Yuen MF, Seto WK, Fung J, Wong DK, Yuen JC, Lai CL. Three years of continuous entecavir therapy in treatment-naïve chronic hepatitis B patients: VIRAL suppression, viral resistance, and clinical safety. Am J Gastroenterol. 2011;106:1264–1271. doi: 10.1038/ajg.2011.45. [ DOI ] [ PubMed ] [ Google Scholar ]
- 52. Lee HW, Lee HJ, Hwang JS, Sohn JH, Jang JY, Han KJ, Park JY, Kim DY, Ahn SH, Paik YH, Lee CK, Lee KS, Chon CY, Han KH. Lamivudine maintenance beyond one year after HBeAg seroconversion is a major factor for sustained virologic response in HBeAg-positive chronic hepatitis B. Hepatology. 2010;51:415–421. doi: 10.1002/hep.23323. [ DOI ] [ PubMed ] [ Google Scholar ]
- 53. Azhari H, Frolkis A, Shaheen AA, Israelson H, Pinto J, Congly SE, Borman MA, Aspinall A, Stinton LM, Swain , Burak MG. Real World Single Center Experience On The Efficacy Of Stopping Long Term Nucleos (T) Ide Analog Therapy In Patients With Chronic Hepatitis B. The Liver Meeting Digital Experience. 2020 [ Google Scholar ]
- 54. Terrault N, Lok AS, Wahed A, Wong DK, Khalili M, Fried MW, Lau D, Ghany MG, Sterling RK, Bisceglie D, Perrillo AM. Randomized trila of 190 weeks of tenmfovir with or without peginterferon alfa for the first 24 weeks followed by protocolozed adults with chronic hepatitis B. The Liver Meeting Digital Experience. 2020 [ Google Scholar ]
- 55. Ramanan V, Shlomai A, Cox DB, Schwartz RE, Michailidis E, Bhatta A, Scott DA, Zhang F, Rice CM, Bhatia SN. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci Rep. 2015;5:10833. doi: 10.1038/srep10833. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 56. Li H, Sheng C, Wang S, Yang L, Liang Y, Huang Y, Liu H, Li P, Yang C, Yang X, Jia L, Xie J, Wang L, Hao R, Du X, Xu D, Zhou J, Li M, Sun Y, Tong Y, Li Q, Qiu S, Song H. Removal of Integrated Hepatitis B Virus DNA Using CRISPR-Cas9. Front Cell Infect Microbiol. 2017;7:91. doi: 10.3389/fcimb.2017.00091. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 57. Yang YC, Chen YH, Kao JH, Ching C, Liu IJ, Wang CC, Tsai CH, Wu FY, Liu CJ, Chen PJ, Chen DS, Yang HC. Permanent Inactivation of HBV Genomes by CRISPR/Cas9-Mediated Non-cleavage Base Editing. Mol Ther Nucleic Acids. 2020;20:480–490. doi: 10.1016/j.omtn.2020.03.005. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 58. Song J, Zhang X, Ge Q, Yuan C, Chu L, Liang HF, Liao Z, Liu Q, Zhang Z, Zhang B. CRISPR/Cas9-mediated knockout of HBsAg inhibits proliferation and tumorigenicity of HBV-positive hepatocellular carcinoma cells. J Cell Biochem. 2018;119:8419–8431. doi: 10.1002/jcb.27050. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 59. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 60. Schluep T, Lickliter J, Hamilton J, Lewis DL, Lai CL, Lau JY, Locarnini SA, Gish RG, Given BD. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin Pharmacol Drug Dev. 2017;6:350–362. doi: 10.1002/cpdd.318. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 61. Wooddell CI, Yuen MF, Chan HL, Gish RG, Locarnini SA, Chavez D, Ferrari C, Given BD, Hamilton J, Kanner SB, Lai CL, Lau JYN, Schluep T, Xu Z, Lanford RE, Lewis DL. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aan0241. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 62. Yuen MF, Schiefke I, Yoon JH, Ahn SH, Heo J, Kim JH, Lik Yuen Chan H, Yoon KT, Klinker H, Manns M, Petersen J, Schluep T, Hamilton J, Given BD, Ferrari C, Lai CL, Locarnini SA, Gish RG. RNA Interference Therapy With ARC-520 Results in Prolonged Hepatitis B Surface Antigen Response in Patients With Chronic Hepatitis B Infection. Hepatology. 2020;72:19–31. doi: 10.1002/hep.31008. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 63. Yuen , M F., Berliba, E., Kim, Y.J., Holmes, J.A., Lim, Y.S., Strasser, S.I., Schwabe, C., Jucov, A., Lee, A.C., Thi, Harasym EP and, T. SAFETY AND PHARMACODYNAMICS OF THE GALNAC-siRNA AB-729 IN SUBJECTS WITH CHRONIC HEPATITIS B INFECTION. The Liver Meeting Digital Experience. 2020 [ Google Scholar ]
- 64. Billioud G, Kruse RL, Carrillo M, Whitten-Bauer C, Gao D, Kim A, Chen L, McCaleb ML, Crosby JR, Hamatake R, Hong Z, Garaigorta U, Swayze E, Bissig KD, Wieland S. In vivo reduction of hepatitis B virus antigenemia and viremia by antisense oligonucleotides. J Hepatol. 2016;64:781–789. doi: 10.1016/j.jhep.2015.11.032. [ DOI ] [ PubMed ] [ Google Scholar ]
- 65. Han K, Cremer J, Elston R, Oliver S, Baptiste-Brown S, Chen S, Gardiner D, Davies M, Saunders J, Hamatake R, Losos J, Leivers M, Hood S, van der Berg F, Paff M, Ritter JM, Theodore D. A Randomized, Double-Blind, Placebo-Controlled, First-Time-in-Human Study to Assess the Safety, Tolerability, and Pharmacokinetics of Single and Multiple Ascending Doses of GSK3389404 in Healthy Subjects. Clin Pharmacol Drug Dev. 2019;8:790–801. doi: 10.1002/cpdd.670. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 66. Yuen M-F, Heo J, Jang JW, Yoon J-H, Kweon YO, Park S-J, Bennett CF, Kwoh TJ. AS067 Hepatitis B virus (HBV) surface antigen (HBsAg) inhibition with isis 505358 in chronic hepatitis B (CHB) patients on stable nucleos (t)ide analogue (NA) regimen and in NA-naive CHB patients: phase 2a, randomized, double-blind, placebo-controlled study. J Hepatol. 2020;73:S49–S50. [ Google Scholar ]
- 67. Hong J, Tan H, Lin T-I, Kang H, Nie Y, Bhattacharya A, Pandey R, Blatt LM, Symons JA, Beigelman and LN. COMBINATION OF ANTISENSE OLIGONUCLEOTIDES (ASOS) ALG-020572 AND ALG-020576 AGAINST HEPATITIS B VIRUS (HBV) IMPROVES ACTIVITY AND CAN BE COMBINED WITH OTHER ANTI-HBV AGENTS. The Liver Meeting Digital Experience. 2020 [ Google Scholar ]
- 68. Bertoletti A. ImmTAV, a New Immunotherapy Targeting the Source of HBV Infection. Hepatology. 2020;72:1514–1517. doi: 10.1002/hep.31527. [ DOI ] [ PubMed ] [ Google Scholar ]
- 69. Fergusson JR, Wallace Z, Connolly MM, Woon AP, Suckling RJ, Hine DW, Barber C, Bunjobpol W, Choi BS, Crespillo S, Dembek M, Dieckmann N, Donoso J, Godinho LF, Grant T, Howe D, McCully ML, Perot C, Sarkar A, Seifert FU, Singh PK, Stegmann KA, Turner B, Verma A, Walker A, Leonard S, Maini MK, Wiederhold K, Dorrell L, Simmons R, Knox A. Immune-Mobilizing Monoclonal T Cell Receptors Mediate Specific and Rapid Elimination of Hepatitis B-Infected Cells. Hepatology. 2020;72:1528–1540. doi: 10.1002/hep.31503. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 70. Gane E, Verdon DJ, Brooks AE, Gaggar A, Nguyen AH, Subramanian GM, Schwabe C, Dunbar PR. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J Hepatol. 2019;71:900–907. doi: 10.1016/j.jhep.2019.06.028. [ DOI ] [ PubMed ] [ Google Scholar ]
- 71. Krebs K, Böttinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, Jäger C, Schmitt E, Bohne F, Aichler M, Uckert W, Abken H, Heikenwalder M, Knolle P, Protzer U. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–465. doi: 10.1053/j.gastro.2013.04.047. [ DOI ] [ PubMed ] [ Google Scholar ]
- 72. Wisskirchen K, Kah J, Malo A, Asen T, Volz T, Allweiss L, Wettengel JM, Lütgehetmann M, Urban S, Bauer T, Dandri M, Protzer U. T cell receptor grafting allows virological control of Hepatitis B virus infection. J Clin Invest. 2019;129:2932–2945. doi: 10.1172/JCI120228. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 73. Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364:2429–2438. doi: 10.1056/NEJMcp1006613. [ DOI ] [ PubMed ] [ Google Scholar ]
- 74. Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [ DOI ] [ PubMed ] [ Google Scholar ]
- 75. The World Health Organization. Global health sector strategy on viral hepatitis 2016-2021. Towards ending viral hepatitis. 2016. [cited 27 June 2021]. Available from: https://www.who.int/publications/i/item/WHO-HIV-2016.06 .
- 76. Guerra J, Garenne M, Mohamed MK, Fontanet A. HCV burden of infection in Egypt: results from a nationwide survey. J Viral Hepat. 2012;19:560–567. doi: 10.1111/j.1365-2893.2011.01576.x. [ DOI ] [ PubMed ] [ Google Scholar ]
- 77. Hofmeister MG, Rosenthal EM, Barker LK, Rosenberg ES, Barranco MA, Hall EW, Edlin BR, Mermin J, Ward JW, Ryerson AB. Estimating Prevalence of Hepatitis C Virus Infection in the United States, 2013-2016. Hepatology. 2019;69:1020–1031. doi: 10.1002/hep.30297. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 78. Frank C, Mohamed MK, Strickland GT, Lavanchy D, Arthur RR, Magder LS, El Khoby T, Abdel-Wahab Y, Aly Ohn ES, Anwar W, Sallam I. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet. 2000;355:887–891. doi: 10.1016/s0140-6736(99)06527-7. [ DOI ] [ PubMed ] [ Google Scholar ]
- 79. Waked I, Esmat G, Elsharkawy A, El-Serafy M, Abdel-Razek W, Ghalab R, Elshishiney G, Salah A, Abdel Megid S, Kabil K, El-Sayed MH, Dabbous H, El Shazly Y, Abo Sliman M, Abou Hashem K, Abdel Gawad S, El Nahas N, El Sobky A, El Sonbaty S, El Tabakh H, Emad E, Gemeah H, Hashem A, Hassany M, Hefnawy N, Hemida AN, Khadary A, Labib K, Mahmoud F, Mamoun S, Marei T, Mekky S, Meshref A, Othman A, Ragab O, Ramadan E, Rehan A, Saad T, Saeed R, Sharshar M, Shawky H, Shawky M, Shehata W, Soror H, Taha M, Talha M, Tealaab A, Zein M, Hashish A, Cordie A, Omar Y, Kamal E, Ammar I, AbdAlla M, El Akel W, Doss W, Zaid H. Screening and Treatment Program to Eliminate Hepatitis C in Egypt. N Engl J Med. 2020;382:1166–1174. doi: 10.1056/NEJMsr1912628. [ DOI ] [ PubMed ] [ Google Scholar ]
- 80. Edlin BR, Eckhardt BJ, Shu MA, Holmberg SD, Swan T. Toward a more accurate estimate of the prevalence of hepatitis C in the United States. Hepatology. 2015;62:1353–1363. doi: 10.1002/hep.27978. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 81. Zibbell JE, Iqbal K, Patel RC, Suryaprasad A, Sanders KJ, Moore-Moravian L, Serrecchia J, Blankenship S, Ward JW, Holtzman D Centers for Disease Control and Prevention (CDC) Increases in hepatitis C virus infection related to injection drug use among persons aged ≤30 years - Kentucky, Tennessee, Virginia, and West Virginia, 2006-2012. MMWR Morb Mortal Wkly Rep. 2015;64:453–458. [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 82. Murphy EL, Bryzman SM, Glynn SA, Ameti DI, Thomson RA, Williams AE, Nass CC, Ownby HE, Schreiber GB, Kong F, Neal KR, Nemo GJ. Risk factors for hepatitis C virus infection in United States blood donors. NHLBI Retrovirus Epidemiology Donor Study (REDS) Hepatology. 2000;31:756–762. doi: 10.1002/hep.510310329. [ DOI ] [ PubMed ] [ Google Scholar ]
- 83. Stramer SL, Glynn SA, Kleinman SH, Strong DM, Caglioti S, Wright DJ, Dodd RY, Busch MP National Heart, Lung, and Blood Institute Nucleic Acid Test Study Group. Detection of HIV-1 and HCV infections among antibody-negative blood donors by nucleic acid-amplification testing. N Engl J Med. 2004;351:760–768. doi: 10.1056/NEJMoa040085. [ DOI ] [ PubMed ] [ Google Scholar ]
- 84. Paintsil E, He H, Peters C, Lindenbach BD, Heimer R. Survival of hepatitis C virus in syringes: implication for transmission among injection drug users. J Infect Dis. 2010;202:984–990. doi: 10.1086/656212. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 85. Mathers BM, Degenhardt L, Phillips B, Wiessing L, Hickman M, Strathdee SA, Wodak A, Panda S, Tyndall M, Toufik A, Mattick RP 2007 Reference Group to the UN on HIV and Injecting Drug Use. Global epidemiology of injecting drug use and HIV among people who inject drugs: a systematic review. Lancet. 2008;372:1733–1745. doi: 10.1016/S0140-6736(08)61311-2. [ DOI ] [ PubMed ] [ Google Scholar ]
- 86. Suryaprasad AG, White JZ, Xu F, Eichler BA, Hamilton J, Patel A, Hamdounia SB, Church DR, Barton K, Fisher C, Macomber K, Stanley M, Guilfoyle SM, Sweet K, Liu S, Iqbal K, Tohme R, Sharapov U, Kupronis BA, Ward JW, Holmberg SD. Emerging epidemic of hepatitis C virus infections among young nonurban persons who inject drugs in the United States, 2006-2012. Clin Infect Dis. 2014;59:1411–1419. doi: 10.1093/cid/ciu643. [ DOI ] [ PubMed ] [ Google Scholar ]
- 87. Farci P, Alter HJ, Shimoda A, Govindarajan S, Cheung LC, Melpolder JC, Sacher RA, Shih JW, Purcell RH. Hepatitis C virus-associated fulminant hepatic failure. N Engl J Med. 1996;335:631–634. doi: 10.1056/NEJM199608293350904. [ DOI ] [ PubMed ] [ Google Scholar ]
- 88. Mosley JW, Operskalski EA, Tobler LH, Andrews WW, Phelps B, Dockter J, Giachetti C, Busch MP. Viral and host factors in early hepatitis C virus infection. Hepatology. 2005;42:86–92. doi: 10.1002/hep.20742. [ DOI ] [ PubMed ] [ Google Scholar ]
- 89. Tillmann HL, Thompson AJ, Patel K, Wiese M, Tenckhoff H, Nischalke HD, Lokhnygina Y, Kullig U, Göbel U, Capka E, Wiegand J, Schiefke I, Güthoff W, Grüngreiff K, König I, Spengler U, McCarthy J, Shianna KV, Goldstein DB, McHutchison JG, Timm J, Nattermann J German Anti-D Study Group. A polymorphism near IL28B is associated with spontaneous clearance of acute hepatitis C virus and jaundice. Gastroenterology. 2010;139:1586–1592, 1592.e1. doi: 10.1053/j.gastro.2010.07.005. [ DOI ] [ PubMed ] [ Google Scholar ]
- 90. Grebely J, Page K, Sacks-Davis R, van der Loeff MS, Rice TM, Bruneau J, Morris MD, Hajarizadeh B, Amin J, Cox AL, Kim AY, McGovern BH, Schinkel J, George J, Shoukry NH, Lauer GM, Maher L, Lloyd AR, Hellard M, Dore GJ, Prins M InC3 Study Group. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology. 2014;59:109–120. doi: 10.1002/hep.26639. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 91. Thein HH, Yi Q, Dore GJ, Krahn MD. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infection: a meta-analysis and meta-regression. Hepatology. 2008;48:418–431. doi: 10.1002/hep.22375. [ DOI ] [ PubMed ] [ Google Scholar ]
- 92. Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol. 2013;10:553–562. doi: 10.1038/nrgastro.2013.107. [ DOI ] [ PubMed ] [ Google Scholar ]
- 93. Desai A, Sandhu S, Lai JP, Sandhu DS. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J Hepatol. 2019;11:1–18. doi: 10.4254/wjh.v11.i1.1. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 94. US Preventive Services Task Force; Owens DK, Davidson KW, Krist AH, Barry MJ, Cabana M, Caughey AB, Donahue K, Doubeni CA, Epling JW Jr, Kubik M, Ogedegbe G, Pbert L, Silverstein M, Simon MA, Tseng CW, Wong JB. Screening for Hepatitis C Virus Infection in Adolescents and Adults: US Preventive Services Task Force Recommendation Statement. JAMA. 2020;323:970–975. doi: 10.1001/jama.2020.1123. [ DOI ] [ PubMed ] [ Google Scholar ]
- 95. Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, Lawitz E, Lok AS, Hinestrosa F, Thuluvath PJ, Schwartz H, Nelson DR, Everson GT, Eley T, Wind-Rotolo M, Huang SP, Gao M, Hernandez D, McPhee F, Sherman D, Hindes R, Symonds W, Pasquinelli C, Grasela DM AI444040 Study Group. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370:211–221. doi: 10.1056/NEJMoa1306218. [ DOI ] [ PubMed ] [ Google Scholar ]
- 96. Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, Shiffman ML, Schiff E, Ghalib R, Ryan M, Rustgi V, Chojkier M, Herring R, Di Bisceglie AM, Pockros PJ, Subramanian GM, An D, Svarovskaia E, Hyland RH, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Pound D, Fried MW ION-3 Investigators. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370:1879–1888. doi: 10.1056/NEJMoa1402355. [ DOI ] [ PubMed ] [ Google Scholar ]
- 97. Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, Romero-Gomez M, Zarski JP, Agarwal K, Buggisch P, Foster GR, Bräu N, Buti M, Jacobson IM, Subramanian GM, Ding X, Mo H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Mangia A, Marcellin P ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370:1889–1898. doi: 10.1056/NEJMoa1402454. [ DOI ] [ PubMed ] [ Google Scholar ]
- 98. Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368:1878–1887. doi: 10.1056/NEJMoa1214853. [ DOI ] [ PubMed ] [ Google Scholar ]
- 99. Solomon SS, Wagner-Cardoso S, Smeaton L, Sowah LA, Wimbish C, Robbins G, Brates I, Scello C, Son A, Avihingsanon A, Linas B, Anthony D, Nunes EP, Kliemann DA, Supparatpinyo K, Kityo C, Tebas P, Bennet JA, Santana-Bagur J, Benson CA, Van Schalkwyk M, Cheinquer N, Naggie S, Wyles D, Sulkowski M. A minimal monitoring approach for the treatment of hepatitis C virus infection (ACTG A5360 [MINMON]): a phase 4, open-label, single-arm trial. The Lancet Gastroenterology & Hepatology. 2022 doi: 10.1016/S2468-1253(21)00397-6. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 100. Litwin AH, Jost J, Wagner K, Heo M, Karasz A, Feinberg J, Kim AY, Lum PJ, Mehta SH, Taylor LE, Tsui JI, Pericot-Valverde I, Page K HERO Study Group. Rationale and design of a randomized pragmatic trial of patient-centered models of hepatitis C treatment for people who inject drugs: The HERO study. Contemp Clin Trials. 2019;87:105859. doi: 10.1016/j.cct.2019.105859. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 101. Pépin J, Abou Chakra CN, Pépin E, Nault V, Valiquette L. Evolution of the global burden of viral infections from unsafe medical injections, 2000-2010. PLoS One. 2014;9:e99677. doi: 10.1371/journal.pone.0099677. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 102. Bailey JR, Barnes E, Cox AL. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology. 2019;156:418–430. doi: 10.1053/j.gastro.2018.08.060. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 103. Micallef JM, Kaldor JM, Dore GJ. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat. 2006;13:34–41. doi: 10.1111/j.1365-2893.2005.00651.x. [ DOI ] [ PubMed ] [ Google Scholar ]
- 104. Osburn WO, Fisher BE, Dowd KA, Urban G, Liu L, Ray SC, Thomas DL, Cox AL. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology. 2010;138:315–324. doi: 10.1053/j.gastro.2009.09.017. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 105. Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, Shapiro M, Alter HJ, Purcell RH. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 106. Bukh J, Engle RE, Faulk K, Wang RY, Farci P, Alter HJ, Purcell RH. Immunoglobulin with High-Titer In Vitro Cross-Neutralizing Hepatitis C Virus Antibodies Passively Protects Chimpanzees from Homologous, but Not Heterologous, Challenge. J Virol. 2015;89:9128–9132. doi: 10.1128/JVI.01194-15. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 107. de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, Gerges S, Vega K, Labitt RN, Donovan BM, Giang E, Krishnan A, Chiriboga L, Charlton MR, Burton DR, Baltimore D, Law M, Rice CM, Ploss A. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med. 2014;6:254ra129. doi: 10.1126/scitranslmed.3009512. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 108. Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med. 2008;14:25–27. doi: 10.1038/nm1698. [ DOI ] [ PubMed ] [ Google Scholar ]
- 109. Law JL, Chen C, Wong J, Hockman D, Santer DM, Frey SE, Belshe RB, Wakita T, Bukh J, Jones CT, Rice CM, Abrignani S, Tyrrell DL, Houghton M. A hepatitis C virus (HCV) vaccine comprising envelope glycoproteins gpE1/gpE2 derived from a single isolate elicits broad cross-genotype neutralizing antibodies in humans. PLoS One. 2013;8:e59776. doi: 10.1371/journal.pone.0059776. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 110. Werbel WA, Durand CM. Pro: Use of Hepatitis C Virus-Positive Donors Should Be Considered Standard of Care. Clin Liver Dis (Hoboken) 2018;12:100–104. doi: 10.1002/cld.743. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 111. Pereira BJ, Milford EL, Kirkman RL, Levey AS. Transmission of hepatitis C virus by organ transplantation. N Engl J Med. 1991;325:454–460. doi: 10.1056/NEJM199108153250702. [ DOI ] [ PubMed ] [ Google Scholar ]
- 112. Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology. 2002;122:889–896. doi: 10.1053/gast.2002.32418. [ DOI ] [ PubMed ] [ Google Scholar ]
- 113. Verna EC, Abdelmessih R, Salomao MA, Lefkowitch J, Moreira RK, Brown RS Jr. Cholestatic hepatitis C following liver transplantation: an outcome-based histological definition, clinical predictors, and prognosis. Liver Transpl. 2013;19:78–88. doi: 10.1002/lt.23559. [ DOI ] [ PubMed ] [ Google Scholar ]
- 114. Gasink LB, Blumberg EA, Localio AR, Desai SS, Israni AK, Lautenbach E. Hepatitis C virus seropositivity in organ donors and survival in heart transplant recipients. JAMA. 2006;296:1843–1850. doi: 10.1001/jama.296.15.1843. [ DOI ] [ PubMed ] [ Google Scholar ]
- 115. Haji SA, Starling RC, Avery RK, Mawhorter S, Tuzcu EM, Schoenhagen P, Cook DJ, Ratliff NB, McCarthy PM, Young JB, Yamani MH. Donor hepatitis-C seropositivity is an independent risk factor for the development of accelerated coronary vasculopathy and predicts outcome after cardiac transplantation. J Heart Lung Transplant. 2004;23:277–283. doi: 10.1016/S1053-2498(03)00148-7. [ DOI ] [ PubMed ] [ Google Scholar ]
- 116. Heo NY, Mannalithara A, Kim D, Udompap P, Tan JC, Kim WR. Long-term Patient and Graft Survival of Kidney Transplant Recipients With Hepatitis C Virus Infection in the United States. Transplantation. 2018;102:454–460. doi: 10.1097/TP.0000000000001953. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 117. Cohen JB, Eddinger KC, Shelton B, Locke JE, Forde KA, Sawinski D. Effect of kidney donor hepatitis C virus serostatus on renal transplant recipient and allograft outcomes. Clin Kidney J. 2017;10:564–572. doi: 10.1093/ckj/sfx048. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 118. Bowring MG, Kucirka LM, Massie AB, Luo X, Cameron A, Sulkowski M, Rakestraw K, Gurakar A, Kuo I, Segev DL, Durand CM. Changes in Utilization and Discard of Hepatitis C-Infected Donor Livers in the Recent Era. Am J Transplant. 2017;17:519–527. doi: 10.1111/ajt.13976. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 119. Kucirka LM, Singer AL, Ros RL, Montgomery RA, Dagher NN, Segev DL. Underutilization of hepatitis C-positive kidneys for hepatitis C-positive recipients. Am J Transplant. 2010;10:1238–1246. doi: 10.1111/j.1600-6143.2010.03091.x. [ DOI ] [ PubMed ] [ Google Scholar ]
- 120. Reese PP, Abt PL, Blumberg EA, Goldberg DS. Transplanting Hepatitis C-Positive Kidneys. N Engl J Med. 2015;373:303–305. doi: 10.1056/NEJMp1505074. [ DOI ] [ PubMed ] [ Google Scholar ]
- 121. Madan S, Patel SR, Rahgozar K, Saeed O, Murthy S, Vukelic S, Sims DB, Shin JJ, Goldstein DJ, Jorde UP. Utilization rates and clinical outcomes of hepatitis C positive donor hearts in the contemporary era. J Heart Lung Transplant. 2019;38:907–917. doi: 10.1016/j.healun.2019.06.023. [ DOI ] [ PubMed ] [ Google Scholar ]
- 122. Durand CM, Bowring MG, Thomas AG, Kucirka LM, Massie AB, Cameron A, Desai NM, Sulkowski M, Segev DL. The Drug Overdose Epidemic and Deceased-Donor Transplantation in the United States: A National Registry Study. Ann Intern Med. 2018;168:702–711. doi: 10.7326/M17-2451. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 123. Durand CM, Bowring MG, Brown DM, Chattergoon MA, Massaccesi G, Bair N, Wesson R, Reyad A, Naqvi FF, Ostrander D, Sugarman J, Segev DL, Sulkowski M, Desai NM. Direct-Acting Antiviral Prophylaxis in Kidney Transplantation From Hepatitis C Virus-Infected Donors to Noninfected Recipients: An Open-Label Nonrandomized Trial. Ann Intern Med. 2018;168:533–540. doi: 10.7326/M17-2871. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 124. Goldberg DS, Abt PL, Blumberg EA, Van Deerlin VM, Levine M, Reddy KR, Bloom RD, Nazarian SM, Sawinski D, Porrett P, Naji A, Hasz R, Suplee L, Trofe-Clark J, Sicilia A, McCauley M, Farooqi M, Gentile C, Smith J, Reese PP. Trial of Transplantation of HCV-Infected Kidneys into Uninfected Recipients. N Engl J Med. 2017;376:2394–2395. doi: 10.1056/NEJMc1705221. [ DOI ] [ PubMed ] [ Google Scholar ]
- 125. Schlendorf KH, Zalawadiya S, Shah AS, Wigger M, Chung CY, Smith S, Danter M, Choi CW, Keebler ME, Brinkley DM, Sacks SB, Ooi H, Perri R, Awad JA, Lewis S, Hayes R, O'Dell H, Darragh C, Carver A, Edmonds C, Ruzevich-Scholl S, Lindenfeld J. Early outcomes using hepatitis C-positive donors for cardiac transplantation in the era of effective direct-acting anti-viral therapies. J Heart Lung Transplant. 2018;37:763–769. doi: 10.1016/j.healun.2018.01.1293. [ DOI ] [ PubMed ] [ Google Scholar ]
- 126. Woolley AE, Singh SK, Goldberg HJ, Mallidi HR, Givertz MM, Mehra MR, Coppolino A, Kusztos AE, Johnson ME, Chen K, Haddad EA, Fanikos J, Harrington DP, Camp PC, Baden LR DONATE HCV Trial Team. Heart and Lung Transplants from HCV-Infected Donors to Uninfected Recipients. N Engl J Med. 2019;380:1606–1617. doi: 10.1056/NEJMoa1812406. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 127. Schlendorf KH, Zalawadiya S, Shah AS, Perri R, Wigger M, Brinkley DM, Danter MR, Menachem JN, Punnoose LR, Balsara K, Sacks SB, Ooi H, Awad JA, Sandhaus E, Schwartz C, O'Dell H, Carver AB, Edmonds CL, Ruzevich-Scholl S, Lindenfeld J. Expanding Heart Transplant in the Era of Direct-Acting Antiviral Therapy for Hepatitis C. JAMA Cardiol. 2020;5:167–174. doi: 10.1001/jamacardio.2019.4748. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 128. Cotter TG, Paul S, Sandıkçı B, Couri T, Bodzin AS, Little EC, Sundaram V, Charlton M. Increasing Utilization and Excellent Initial Outcomes Following Liver Transplant of Hepatitis C Virus (HCV)-Viremic Donors Into HCV-Negative Recipients: Outcomes Following Liver Transplant of HCV-Viremic Donors. Hepatology. 2019;69:2381–2395. doi: 10.1002/hep.30540. [ DOI ] [ PubMed ] [ Google Scholar ]
- 129. Couri T, Katz J, Stoeckle K, Nugooru A, Yeh H, Chung R, Paul S. Provider Attitudes toward the Use of Hepatitis C Virus-Positive Organs in Kidney Transplantation. Am J Nephrol. 2019;50:168–176. doi: 10.1159/000502049. [ DOI ] [ PubMed ] [ Google Scholar ]
- 130. Rizzetto M, Canese MG, Aricò S, Crivelli O, Trepo C, Bonino F, Verme G. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. 1977;18:997–1003. doi: 10.1136/gut.18.12.997. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 131. Hadziyannis SJ. Review: hepatitis delta. J Gastroenterol Hepatol. 1997;12:289–298. doi: 10.1111/j.1440-1746.1997.tb00424.x. [ DOI ] [ PubMed ] [ Google Scholar ]
- 132. Farci P, Anna Niro G. Current and Future Management of Chronic Hepatitis D. Gastroenterol Hepatol (N Y) 2018;14:342–351. [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 133. Stockdale AJ, Kreuels B, Henrion MYR, Giorgi E, Kyomuhangi I, de Martel C, Hutin Y, Geretti AM. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J Hepatol. 2020;73:523–532. doi: 10.1016/j.jhep.2020.04.008. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 134. Lai MM. The molecular biology of hepatitis delta virus. Annu Rev Biochem. 1995;64:259–286. doi: 10.1146/annurev.bi.64.070195.001355. [ DOI ] [ PubMed ] [ Google Scholar ]
- 135. Popper H, Thung SN, Gerber MA, Hadler SC, de Monzon M, Ponzetto A, Anzola E, Rivera D, Mondolfi A, Bracho A. Histologic studies of severe delta agent infection in Venezuelan Indians. Hepatology. 1983;3:906–912. doi: 10.1002/hep.1840030603. [ DOI ] [ PubMed ] [ Google Scholar ]
- 136. Kamimura T, Ponzetto A, Bonino F, Feinstone SM, Gerin JL, Purcell RH. Cytoplasmic tubular structures in liver of HBsAg carrier chimpanzees infected with delta agent and comparison with cytoplasmic structures in non-A, non-B hepatitis. Hepatology. 1983;3:631–637. doi: 10.1002/hep.1840030502. [ DOI ] [ PubMed ] [ Google Scholar ]
- 137. Cole SM, Gowans EJ, Macnaughton TB, Hall PD, Burrell CJ. Direct evidence for cytotoxicity associated with expression of hepatitis delta virus antigen. Hepatology. 1991;13:845–851. [ PubMed ] [ Google Scholar ]
- 138. Guilhot S, Huang SN, Xia YP, La Monica N, Lai MM, Chisari FV. Expression of the hepatitis delta virus large and small antigens in transgenic mice. J Virol. 1994;68:1052–1058. doi: 10.1128/jvi.68.2.1052-1058.1994. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 139. Verme G, Amoroso P, Lettieri G, Pierri P, David E, Sessa F, Rizzi R, Bonino F, Recchia S, Rizzetto M. A histological study of hepatitis delta virus liver disease. Hepatology. 1986;6:1303–1307. doi: 10.1002/hep.1840060613. [ DOI ] [ PubMed ] [ Google Scholar ]
- 140. Samuel D, Zignego A, Reynes M, Ferry C, Arulnaden JL, David M-F, Gigou M, Bismuth A, Mathieu D, Gentilini P, Benhamou J, Brechot C, Bismuth H. Long-term clinical and virological outcome after liver transplantation for cirrhosis caused by chronic delta hepatitis. Hepatology. 1995;21:333–339. [ PubMed ] [ Google Scholar ]
- 141. Tassopoulos NC, Koutelou MG, Macagno S, Zorbas P, Rizzetto M. Diagnostic significance of IgM antibody to hepatitis delta virus in fulminant hepatitis B. J Med Virol. 1990;30:174–177. doi: 10.1002/jmv.1890300305. [ DOI ] [ PubMed ] [ Google Scholar ]
- 142. Abbas Z, Afzal R. Life cycle and pathogenesis of hepatitis D virus: A review. World J Hepatol. 2013;5:666–675. doi: 10.4254/wjh.v5.i12.666. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 143. Fattovich G, Giustina G, Christensen E, Pantalena M, Zagni I, Realdi G, Schalm SW. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep) Gut. 2000;46:420–426. doi: 10.1136/gut.46.3.420. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 144. Romeo R, Del Ninno E, Rumi M, Russo A, Sangiovanni A, de Franchis R, Ronchi G, Colombo M. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136:1629–1638. doi: 10.1053/j.gastro.2009.01.052. [ DOI ] [ PubMed ] [ Google Scholar ]
- 145. Rizzetto M. Hepatitis D Virus: Introduction and Epidemiology. Cold Spring Harb Perspect Med. 2015;5:a021576. doi: 10.1101/cshperspect.a021576. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 146. Asselah T, Loureiro D, Tout I, Castelnau C, Boyer N, Marcellin P, Mansouri A. Future treatments for hepatitis delta virus infection. Liver Int. 2020;40 Suppl 1:54–60. doi: 10.1111/liv.14356. [ DOI ] [ PubMed ] [ Google Scholar ]
- 147. Farci P, Mandas A, Coiana A, Lai ME, Desmet V, Van Eyken P, Gibo Y, Caruso L, Scaccabarozzi S, Criscuolo D. Treatment of chronic hepatitis D with interferon alfa-2a. N Engl J Med. 1994;330:88–94. doi: 10.1056/NEJM199401133300202. [ DOI ] [ PubMed ] [ Google Scholar ]
- 148. Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, Scioscia R, Serra G, Lai ME, Loy M, Caruso L, Desmet V, Purcell RH, Balestrieri A. Long-term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology. 2004;126:1740–1749. doi: 10.1053/j.gastro.2004.03.017. [ DOI ] [ PubMed ] [ Google Scholar ]
- 149. Erhardt A, Gerlich W, Starke C, Wend U, Donner A, Sagir A, Heintges T, Häussinger D. Treatment of chronic hepatitis delta with pegylated interferon-alpha2b. Liver Int. 2006;26:805–810. doi: 10.1111/j.1478-3231.2006.01279.x. [ DOI ] [ PubMed ] [ Google Scholar ]
- 150. Castelnau C, Le Gal F, Ripault MP, Gordien E, Martinot-Peignoux M, Boyer N, Pham BN, Maylin S, Bedossa P, Dény P, Marcellin P, Gault E. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: relevance of quantitative RT-PCR for follow-up. Hepatology. 2006;44:728–735. doi: 10.1002/hep.21325. [ DOI ] [ PubMed ] [ Google Scholar ]
- 151. Kabaçam G, Onder FO, Yakut M, Seven G, Karatayli SC, Karatayli E, Savas B, Idilman R, Bozdayi AM, Yurdaydin C. Entecavir treatment of chronic hepatitis D. Clin Infect Dis. 2012;55:645–650. doi: 10.1093/cid/cis459. [ DOI ] [ PubMed ] [ Google Scholar ]
- 152. James G, Sidhu P, Raza M. First report of successful clearance of hepatitis B and D coinfection with tenofovir monotherapy. Hepatology. 2015;62:317–318. doi: 10.1002/hep.27446. [ DOI ] [ PubMed ] [ Google Scholar ]
- 153. Soriano V, Vispo E, Sierra-Enguita R, Mendoza Cd, Fernández-Montero JV, Labarga P, Barreiro P. Efficacy of prolonged tenofovir therapy on hepatitis delta in HIV-infected patients. AIDS. 2014;28:2389–2394. doi: 10.1097/QAD.0000000000000417. [ DOI ] [ PubMed ] [ Google Scholar ]
- 154. Wedemeyer H, Yurdaydin C, Hardtke S, Caruntu FA, Curescu MG, Yalcin K, Akarca US, Gürel S, Zeuzem S, Erhardt A, Lüth S, Papatheodoridis GV, Keskin O, Port K, Radu M, Celen MK, Idilman R, Weber K, Stift J, Wittkop U, Heidrich B, Mederacke I, von der Leyen H, Dienes HP, Cornberg M, Koch A, Manns MP HIDIT-II study team. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): a randomised, placebo controlled, phase 2 trial. Lancet Infect Dis. 2019;19:275–286. doi: 10.1016/S1473-3099(18)30663-7. [ DOI ] [ PubMed ] [ Google Scholar ]
- 155. Wedemeyer H, Yurdaydìn C, Dalekos GN, Erhardt A, Çakaloğlu Y, Değertekin H, Gürel S, Zeuzem S, Zachou K, Bozkaya H, Koch A, Bock T, Dienes HP, Manns MP HIDIT Study Group. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med. 2011;364:322–331. doi: 10.1056/NEJMoa0912696. [ DOI ] [ PubMed ] [ Google Scholar ]
- 156. Nimgaonkar I, Ding Q, Schwartz RE, Ploss A. Hepatitis E virus: advances and challenges. Nat Rev Gastroenterol Hepatol. 2018;15:96–110. doi: 10.1038/nrgastro.2017.150. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 157. Meng XJ. Expanding Host Range and Cross-Species Infection of Hepatitis E Virus. PLoS Pathog. 2016;12:e1005695. doi: 10.1371/journal.ppat.1005695. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 158. Zhang J, Zhang XF, Huang SJ, Wu T, Hu YM, Wang ZZ, Wang H, Jiang HM, Wang YJ, Yan Q, Guo M, Liu XH, Li JX, Yang CL, Tang Q, Jiang RJ, Pan HR, Li YM, Shih JW, Ng MH, Zhu FC, Xia NS. Long-term efficacy of a hepatitis E vaccine. N Engl J Med. 2015;372:914–922. doi: 10.1056/NEJMoa1406011. [ DOI ] [ PubMed ] [ Google Scholar ]
- 159. The World Health Organization. Waterborne Outbreaks of Hepatitis E: Recognition, Investigation, and Control. 2014. Available from: https://apps.who.int/iris/bitstream/handle/10665/129448/9789241507608_eng.pdf;jsessionid=71621FB7964A3D06CF9019A4157D9963?sequence=1 .
- 160. Balayan MS, Andjaparidze AG, Savinskaya SS, Ketiladze ES, Braginsky DM, Savinov AP, Poleschuk VF. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology. 1983;20:23–31. doi: 10.1159/000149370. [ DOI ] [ PubMed ] [ Google Scholar ]
- 161. Rein DB, Stevens GA, Theaker J, Wittenborn JS, Wiersma ST. The global burden of hepatitis E virus genotypes 1 and 2 in 2005. Hepatology. 2012;55:988–997. doi: 10.1002/hep.25505. [ DOI ] [ PubMed ] [ Google Scholar ]
- 162. Jiménez de Oya N, de Blas I, Blázquez AB, Martín-Acebes MA, Halaihel N, Gironés O, Saiz JC, Escribano-Romero E. Widespread distribution of hepatitis E virus in Spanish pig herds. BMC Res Notes. 2011;4:412. doi: 10.1186/1756-0500-4-412. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 163. Widén F, Sundqvist L, Matyi-Toth A, Metreveli G, Belák S, Hallgren G, Norder H. Molecular epidemiology of hepatitis E virus in humans, pigs and wild boars in Sweden. Epidemiol Infect. 2011;139:361–371. doi: 10.1017/S0950268810001342. [ DOI ] [ PubMed ] [ Google Scholar ]
- 164. Meng XJ, Halbur PG, Shapiro MS, Govindarajan S, Bruna JD, Mushahwar IK, Purcell RH, Emerson SU. Genetic and experimental evidence for cross-species infection by swine hepatitis E virus. J Virol. 1998;72:9714–9721. doi: 10.1128/jvi.72.12.9714-9721.1998. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 165. Meng XJ, Halbur PG, Haynes JS, Tsareva TS, Bruna JD, Royer RL, Purcell RH, Emerson SU. Experimental infection of pigs with the newly identified swine hepatitis E virus (swine HEV), but not with human strains of HEV. Arch Virol. 1998;143:1405–1415. doi: 10.1007/s007050050384. [ DOI ] [ PubMed ] [ Google Scholar ]
- 166. Balayan MS, Usmanov RK, Zamyatina NA, Djumalieva DI, Karas FR. Brief report: experimental hepatitis E infection in domestic pigs. J Med Virol. 1990;32:58–59. doi: 10.1002/jmv.1890320110. [ DOI ] [ PubMed ] [ Google Scholar ]
- 167. Berto A, Grierson S, Hakze-van der Honing R, Martelli F, Johne R, Reetz J, Ulrich RG, Pavio N, Van der Poel WH, Banks M. Hepatitis E virus in pork liver sausage, France. Emerg Infect Dis. 2013;19:264–266. doi: 10.3201/eid1902.121255. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 168. Di Bartolo I, Diez-Valcarce M, Vasickova P, Kralik P, Hernandez M, Angeloni G, Ostanello F, Bouwknegt M, Rodríguez-Lázaro D, Pavlik I, Ruggeri FM. Hepatitis E virus in pork production chain in Czech Republic, Italy, and Spain, 2010. Emerg Infect Dis. 2012;18:1282–1289. doi: 10.3201/eid1808.111783. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 169. Berto A, Martelli F, Grierson S, Banks M. Hepatitis E virus in pork food chain, United Kingdom, 2009-2010. Emerg Infect Dis. 2012;18:1358–1360. doi: 10.3201/eid1808.111647. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 170. Barnaud E, Rogée S, Garry P, Rose N, Pavio N. Thermal inactivation of infectious hepatitis E virus in experimentally contaminated food. Appl Environ Microbiol. 2012;78:5153–5159. doi: 10.1128/AEM.00436-12. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 171. Hewitt PE, Ijaz S, Brailsford SR, Brett R, Dicks S, Haywood B, Kennedy IT, Kitchen A, Patel P, Poh J, Russell K, Tettmar KI, Tossell J, Ushiro-Lumb I, Tedder RS. Hepatitis E virus in blood components: a prevalence and transmission study in southeast England. Lancet. 2014;384:1766–1773. doi: 10.1016/S0140-6736(14)61034-5. [ DOI ] [ PubMed ] [ Google Scholar ]
- 172. Pawlotsky JM. Hepatitis E screening for blood donations: an urgent need? Lancet. 2014;384:1729–1730. doi: 10.1016/S0140-6736(14)61187-9. [ DOI ] [ PubMed ] [ Google Scholar ]
- 173. Hadifar S, Sedighi M, Mostafaei S, Miri A, Amiri H, Abiri R, Babaei F, Kabir K, Moghoofei M. Prevalence of hepatitis E infection in the general population of Iran: a systematic review and meta-analysis. Future Virology. 2017:12. [ Google Scholar ]
- 174. Tavakoli A, Alavian SM, Moghoofei M, Mostafaei S, Abbasi S, Farahmand M. Seroepidemiology of hepatitis E virus infection in patients undergoing maintenance hemodialysis: Systematic review and meta-analysis. Ther Apher Dial. 2021;25:4–15. doi: 10.1111/1744-9987.13507. [ DOI ] [ PubMed ] [ Google Scholar ]
- 175. Suneetha PV, Pischke S, Schlaphoff V, Grabowski J, Fytili P, Gronert A, Bremer B, Markova A, Jaroszewicz J, Bara C, Manns MP, Cornberg M, Wedemeyer H. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology. 2012;55:695–708. doi: 10.1002/hep.24738. [ DOI ] [ PubMed ] [ Google Scholar ]
- 176. Pischke S, Behrendt P, Bock CT, Jilg W, Manns MP, Wedemeyer H. Hepatitis E in Germany--an under-reported infectious disease. Dtsch Arztebl Int. 2014;111:577–583. doi: 10.3238/arztebl.2014.0577. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 177. Aggarwal R, Naik S. Epidemiology of hepatitis E: current status. J Gastroenterol Hepatol. 2009;24:1484–1493. doi: 10.1111/j.1440-1746.2009.05933.x. [ DOI ] [ PubMed ] [ Google Scholar ]
- 178. Pérez-Gracia MT, Suay-García B, Mateos-Lindemann ML. Hepatitis E and pregnancy: current state. Rev Med Virol. 2017;27:e1929. doi: 10.1002/rmv.1929. [ DOI ] [ PubMed ] [ Google Scholar ]
- 179. Neukam K, Barreiro P, Macías J, Avellón A, Cifuentes C, Martín-Carbonero L, Echevarría JM, Vargas J, Soriano V, Pineda JA. Chronic hepatitis E in HIV patients: rapid progression to cirrhosis and response to oral ribavirin. Clin Infect Dis. 2013;57:465–468. doi: 10.1093/cid/cit224. [ DOI ] [ PubMed ] [ Google Scholar ]
- 180. Tavitian S, Péron JM, Huynh A, Mansuy JM, Ysebaert L, Huguet F, Vinel JP, Attal M, Izopet J, Récher C. Hepatitis E virus excretion can be prolonged in patients with hematological malignancies. J Clin Virol. 2010;49:141–144. doi: 10.1016/j.jcv.2010.06.016. [ DOI ] [ PubMed ] [ Google Scholar ]
- 181. Kamar N, Selves J, Mansuy JM, Ouezzani L, Péron JM, Guitard J, Cointault O, Esposito L, Abravanel F, Danjoux M, Durand D, Vinel JP, Izopet J, Rostaing L. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med. 2008;358:811–817. doi: 10.1056/NEJMoa0706992. [ DOI ] [ PubMed ] [ Google Scholar ]
- 182. Kamar N, Abravanel F, Selves J, Garrouste C, Esposito L, Lavayssière L, Cointault O, Ribes D, Cardeau I, Nogier MB, Mansuy JM, Muscari F, Peron JM, Izopet J, Rostaing L. Influence of immunosuppressive therapy on the natural history of genotype 3 hepatitis-E virus infection after organ transplantation. Transplantation. 2010;89:353–360. doi: 10.1097/TP.0b013e3181c4096c. [ DOI ] [ PubMed ] [ Google Scholar ]
- 183. Kamar N, Izopet J, Tripon S, Bismuth M, Hillaire S, Dumortier J, Radenne S, Coilly A, Garrigue V, D'Alteroche L, Buchler M, Couzi L, Lebray P, Dharancy S, Minello A, Hourmant M, Roque-Afonso AM, Abravanel F, Pol S, Rostaing L, Mallet V. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N Engl J Med. 2014;370:1111–1120. doi: 10.1056/NEJMoa1215246. [ DOI ] [ PubMed ] [ Google Scholar ]
- 184. Sinclair SM, Jones JK, Miller RK, Greene MF, Kwo PY, Maddrey WC. The Ribavirin Pregnancy Registry: An Interim Analysis of Potential Teratogenicity at the Mid-Point of Enrollment. Drug Saf. 2017;40:1205–1218. doi: 10.1007/s40264-017-0566-6. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 185. van der Valk M, Zaaijer HL, Kater AP, Schinkel J. Sofosbuvir shows antiviral activity in a patient with chronic hepatitis E virus infection. J Hepatol. 2017;66:242–243. doi: 10.1016/j.jhep.2016.09.014. [ DOI ] [ PubMed ] [ Google Scholar ]
- 186. Haagsma EB, Riezebos-Brilman A, van den Berg AP, Porte RJ, Niesters HG. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon alpha-2b. Liver Transpl. 2010;16:474–477. doi: 10.1002/lt.22014. [ DOI ] [ PubMed ] [ Google Scholar ]
- 187. Reshetnyak VI, Karlovich TI, Ilchenko LU. Hepatitis G virus. World J Gastroenterol. 2008;14:4725–4734. doi: 10.3748/wjg.14.4725. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- 188. Wiwanitkit V. Hepatitis G virus RNA positivity among the voluntary blood donors: a summary. Ann Hepatol. 2005;4:43–46. [ PubMed ] [ Google Scholar ]
- 189. Alter HJ, Nakatsuji Y, Melpolder J, Wages J, Wesley R, Shih JW-K, Kim JP. The Incidence of Transfusion-Associated Hepatitis G Virus Infection and Its Relation to Liver Disease. New England Journal of Medicine. 1997;336:747–754. doi: 10.1056/NEJM199703133361102. [ DOI ] [ PubMed ] [ Google Scholar ]
- 190. Alter HJ. The cloning and clinical implications of HGV and HGBV-C. N Engl J Med. 1996;334:1536–1537. doi: 10.1056/NEJM199606063342310. [ DOI ] [ PubMed ] [ Google Scholar ]
- 191. Masuko K, Mitsui T, Iwano K, Yamazaki C, Okuda K, Meguro T, Murayama N, Inoue T, Tsuda F, Okamoto H, Miyakawa Y, Mayumi M. Infection with hepatitis GB virus C in patients on maintenance hemodialysis. N Engl J Med. 1996;334:1485–1490. doi: 10.1056/NEJM199606063342301. [ DOI ] [ PubMed ] [ Google Scholar ]
- 192. Fried MW, Khudyakov YE, Smallwood GA, Cong M, Nichols B, Diaz E, Siefert P, Gutekunst K, Gordon RD, Boyer TD, Fields HA. Hepatitis G virus co-infection in liver transplantation recipients with chronic hepatitis C and nonviral chronic liver disease. Hepatology. 1997;25:1271–1275. doi: 10.1002/hep.510250536. [ DOI ] [ PubMed ] [ Google Scholar ]
- 193. Pessoa MG, Terrault NA, Detmer J, Kolberg J, Collins M, Hassoba HM, Wright TL. Quantitation of hepatitis G and C viruses in the liver: evidence that hepatitis G virus is not hepatotropic. Hepatology. 1998;27:877–880. doi: 10.1002/hep.510270335. [ DOI ] [ PubMed ] [ Google Scholar ]
- 194. Kanda T, Yokosuka O, Ehata T, Maru Y, Imazeki F, Saisho H, Shiratori Y, Omata M. Detection of GBV-C RNA in patients with non-A-E fulminant hepatitis by reverse-transcription polymerase chain reaction. Hepatology. 1997;25:1261–1265. doi: 10.1002/hep.510250534. [ DOI ] [ PubMed ] [ Google Scholar ]
- 195. Bukh J, Kim JP, Govindarajan S, Apgar CL, Foung SK, Wages J Jr, Yun AJ, Shapiro M, Emerson SU, Purcell RH. Experimental infection of chimpanzees with hepatitis G virus and genetic analysis of the virus. J Infect Dis. 1998;177:855–862. doi: 10.1086/515255. [ DOI ] [ PubMed ] [ Google Scholar ]
- View on publisher site
- PDF (613.5 KB)
- Collections
Similar articles
Cited by other articles, links to ncbi databases.
- Download .nbib .nbib
- Format: AMA APA MLA NLM
Add to Collections
IMAGES
VIDEO
COMMENTS
The clinical presentation of viral hepatitis can be different in each individual, depending on the type of hepatitis virus causing the infection. Patients can be entirely asymptomatic or only mildly symptomatic at presentation.
The clinical presentation of infectious hepatitis varies with the individual, as well as with the specific causative virus. Some patients may be entirely asymptomatic or only mildly...
Clinical Overview of Viral Hepatitis. Key points. Many people with a viral hepatitis infection do not have symptoms and are unaware of their infection. Chronic hepatitis B and chronic hepatitis C can cause serious health problems, including liver damage, cirrhosis, liver cancer, and even death.
The clinical presentation of acute hepatitis depends on the underlying etiology. It can clinically manifest with various clinical signs and symptoms, ranging from asymptomatic elevated liver function tests to acute liver failure requiring liver transplantation.
Acute viral hepatitis is diffuse liver inflammation caused by specific hepatotropic viruses that have diverse modes of transmission and epidemiologies. Although acute viral hepatitis can be asymptomatic, a nonspecific viral prodrome is often followed by anorexia, nausea, and often fever or right upper quadrant pain.
In this review of viral hepatitis infections, we discuss the pertinent clinical information and recent organizational guidelines for each of the individual hepatitis viruses while also synthesizing this information with the latest research to focus on exciting future directions for each virus.