Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Published: 17 February 2022

Spatial organization of transcribed eukaryotic genes
- Susanne Leidescher 1 ,
- Johannes Ribisel 2 na1 ,
- Simon Ullrich 1 na1 ,
- Yana Feodorova 1 , 3 ,
- Erica Hildebrand 4 ,
- Alexandra Galitsyna ORCID: orcid.org/0000-0001-8969-5694 5 ,
- Sebastian Bultmann ORCID: orcid.org/0000-0002-5681-1043 1 ,
- Stephanie Link 6 ,
- Katharina Thanisch 1 nAff8 ,
- Christopher Mulholland ORCID: orcid.org/0000-0001-8981-0111 1 ,
- Job Dekker ORCID: orcid.org/0000-0001-5631-0698 4 , 7 ,
- Heinrich Leonhardt ORCID: orcid.org/0000-0002-5086-6449 1 ,
- Leonid Mirny ORCID: orcid.org/0000-0002-0785-5410 2 &
- Irina Solovei ORCID: orcid.org/0000-0002-6813-7279 1
Nature Cell Biology volume 24 , pages 327–339 ( 2022 ) Cite this article
11k Accesses
30 Citations
63 Altmetric
Metrics details
- Chromatin structure
- Nuclear organization
- Transcription
Despite the well-established role of nuclear organization in the regulation of gene expression, little is known about the reverse: how transcription shapes the spatial organization of the genome. Owing to the small sizes of most previously studied genes and the limited resolution of microscopy, the structure and spatial arrangement of a single transcribed gene are still poorly understood. Here we study several long highly expressed genes and demonstrate that they form open-ended transcription loops with polymerases moving along the loops and carrying nascent RNAs. Transcription loops can span across micrometres, resembling lampbrush loops and polytene puffs. The extension and shape of transcription loops suggest their intrinsic stiffness, which we attribute to decoration with multiple voluminous nascent ribonucleoproteins. Our data contradict the model of transcription factories and suggest that although microscopically resolvable transcription loops are specific for long highly expressed genes, the mechanisms underlying their formation could represent a general aspect of eukaryotic transcription.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Organization and regulation of gene transcription
Patrick Cramer

Nuclear compartmentalization as a mechanism of quantitative control of gene expression
Prashant Bhat, Drew Honson & Mitchell Guttman

The spatial organization of transcriptional control
Antonina Hafner & Alistair Boettiger
Data availability
Hi-C data have been uploaded to Gene Expression Omnibus (GEO) and are available under accession GSE150704 . ChIP–seq and RNA-seq data are available at ArrayExpress (EMBL-EBI) under accession E-MTAB-9060 ( https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-9060/ ). Previously published reference genome mm10 and gene annotation of the C57BL/6 J strain were downloaded from the Ensemble database (version GRCm38, release 74). Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding authors on reasonable request.
Code availability
The used code for the measurement of flank distances is available at https://github.com/hoerldavid/fish_analysis ; code used for the analysis of Hi-C data (Cooler, Cooltools, Distiller, Pairtools) is available at https://github.com/open2c ; the code used for polymer simulations is available at https://github.com/mirnylab/openmm-polymer-legacy .
Andersson, R. & Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21 , 71–87 (2020).
Article CAS PubMed Google Scholar
Cramer, P. Organization and regulation of gene transcription. Nature 573 , 45–54 (2019).
Herzel, L., Straube, K. & Neugebauer, K. M. Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Res. 28 , 1008–1019 (2018).
Article CAS PubMed PubMed Central Google Scholar
Feodorova, Y., Falk, M., Mirny, L. A. & Solovei, I. Viewing nuclear architecture through the eyes of nocturnal mammals. Trends Cell Biol. 30 , 276–289 (2020).
Solovei, I., Thanisch, K. & Feodorova, Y. How to rule the nucleus: divide et impera. Curr. Opin. Cell Biol. 40 , 47–59 (2016).
van Steensel, B. & Belmont, A. S. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169 , 780–791 (2017).
Article PubMed PubMed Central CAS Google Scholar
Macgregor, H. C. An Introduction to Animal Cytogenetics (Chapman & Hall, 1993).
Bjork, P. & Wieslander, L. The Balbiani ring story: synthesis, assembly, processing, and transport of specific messenger RNA–protein complexes. Annu. Rev. Biochem. 84 , 65–92 (2015).
Article PubMed CAS Google Scholar
Chakalova, L. & Fraser, P. Organization of transcription. Cold Spring Harb. Perspect. Biol. 2 , a000729 (2010).
Osborne, C. S. et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36 , 1065–1071 (2004).
Schoenfelder, S. et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 42 , 53–61 (2010).
Cook, P. R. The organization of replication and transcription. Science 284 , 1790–1795 (1999).
Papantonis, A. & Cook, P. R. Transcription factories: genome organization and gene regulation. Chem. Rev. 113 , 8683–8705 (2013).
Mateo, L. J. et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568 , 49–54 (2019).
Nora, E. P. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485 , 381–385 (2012).
Rodermund, L. et al. Time-resolved structured illumination microscopy reveals key principles of Xist RNA spreading. Science 372 , (2021).
Morgan, G. T. Imaging the dynamics of transcription loops in living chromosomes. Chromosoma 127 , 361–374 (2018).
Article PubMed PubMed Central Google Scholar
Schermelleh, L., Heintzmann, R. & Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 190 , 165–175 (2010).
Jjingo, D., Huda, A., Gundapuneni, M., Marino-Ramirez, L. & Jordan, I. K. Effect of the transposable element environment of human genes on gene length and expression. Genome Biol. Evol. 3 , 259–271 (2011).
Hutchison, N. Lampbrush chromosomes of the chicken, Gallus domesticus . J. Cell Biol. 105 , 1493–1500 (1987).
Miller, O. L. & Beatty, B. R. Visualization of nuclear genes. Science 164 , 955–957 (1969).
Article PubMed Google Scholar
Kaufmann, R., Cremer, C. & Gall, J. G. Superresolution imaging of transcription units on newt lampbrush chromosomes. Chromosome Res. 20 , 1009–1015 (2012).
Carmo-Fonseca, M. & Kirchhausen, T. The timing of pre-mRNA splicing visualized in real-time. Nucleus 5 , 11–14 (2014).
Ansari, A. & Hampsey, M. A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 19 , 2969–2978 (2005).
Larsson, A. J. M. et al. Genomic encoding of transcriptional burst kinetics. Nature 565 , 251–254 (2019).
Tantale, K. et al. A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat. Commun. 7 , 12248 (2016).
Chavez, A. et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12 , 326–328 (2015).
Bensaude, O. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2 , 103–108 (2011).
Mahy, N. L., Perry, P. E., Gilchrist, S., Baldock, R. A. & Bickmore, W. A. Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J. Cell Biol. 157 , 579–589 (2002).
Brown, J. M. et al. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J. Cell Biol. 182 , 1083–1097 (2008).
Chambeyron, S., Da Silva, N. R., Lawson, K. A. & Bickmore, W. A. Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development 132 , 2215–2223 (2005).
Mahy, N. L., Perry, P. E. & Bickmore, W. A. Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. J. Cell Biol. 159 , 753–763 (2002).
Volpi, E. V. et al. Large-scale chromatin organization of the major histocompatibility complex and other regions of human chromosome 6 and its response to interferon in interphase nuclei. J. Cell Sci. 113 , 1565–1576 (2000).
Williams, R. R., Broad, S., Sheer, D. & Ragoussis, J. Subchromosomal positioning of the epidermal differentiation complex (EDC) in keratinocyte and lymphoblast interphase nuclei. Exp. Cell. Res. 272 , 163–175 (2002).
Abramo, K. et al. A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat. Cell Biol. 21 , 1393–1402 (2019).
Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F. & Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30 , 90–98 (2012).
Article CAS Google Scholar
Shah, S. et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174 , 363–376 e316 (2018).
Hsieh, T. S. et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol. Cell 78 , 539–553 (2020).
Banigan, E. J. & Mirny, L. A. The interplay between asymmetric and symmetric DNA loop extrusion. eLife 9, (2020).
Brandao, H. B. et al. RNA polymerases as moving barriers to condensin loop extrusion. Proc. Natl Acad. Sci. USA 116 , 20489–20499 (2019).
Banigan, E. J. et al. Transcription shapes 3D chromatin organization by interacting with loop-extruding cohesin complexes. https://doi.org/10.1101/2022.01.07.475367 (2022).
Muller-McNicoll, M. & Neugebauer, K. M. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat. Rev. Genet. 14 , 275–287 (2013).
Olins, A. L. & Olins, D. E. Spheroid chromatin units (v bodies). Science 183 , 330–332 (1974).
Liu, X., Farnung, L., Wigge, C. & Cramer, P. Cryo-EM structure of a mammalian RNA polymerase II elongation complex inhibited by alpha-amanitin. J. Biol. Chem. 293 , 7189–7194 (2018).
Paturej, J., Sheiko, S. S., Panyukov, S. & Rubinstein, M. Molecular structure of bottlebrush polymers in melts. Sci. Adv. 2 , e1601478 (2016).
Kotake, Y. et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 3 , 570–575 (2007).
Mirny, L. A. & Solovei, I. Keeping chromatin in the loop(s). Nat. Rev. Mol. Cell Biol. 22 , 439–440 (2021).
Cho, W. K. et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361 , 412–415 (2018).
Guo, Y. E. et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572 , 543–548 (2019).
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. & Sharp, P. A. A phase separation model for transcriptional control. Cell 169 , 13–23 (2017).
Cisse, I. I. et al. Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341 , 664–667 (2013).
Henninger, J. E. et al. RNA-mediated feedback control of transcriptional condensates. Cell 184 , 207–225 e224 (2021).
Hampsey, M., Singh, B. N., Ansari, A., Laine, J. P. & Krishnamurthy, S. Control of eukaryotic gene expression: gene loops and transcriptional memory. Adv. Enzyme Regul. 51 , 118–125 (2011).
Martin, M., Cho, J., Cesare, A. J., Griffith, J. D. & Attardi, G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 123 , 1227–1240 (2005).
Singh, B. N. & Hampsey, M. A transcription-independent role for TFIIB in gene looping. Mol. Cell 27 , 806–816 (2007).
Lee, K., Hsiung, C. C., Huang, P., Raj, A. & Blobel, G. A. Dynamic enhancer–gene body contacts during transcription elongation. Genes Dev. 29 , 1992–1997 (2015).
Zheng, M. et al. Multiplex chromatin interactions with single-molecule precision. Nature 566 , 558–562 (2019).
Cremer, T. & Cremer, M. Chromosome territories. Cold Spring Harb. Perspect. Biol. 2 , a003889 (2010).
Keizer, V. I. P. et al. Live-cell micromanipulation of a genomic locus reveals interphase chromatin mechanics. Preprint at bioRxiv https://doi.org/10.1101/2021.04.20.439763 (2021).
Khanna, N., Zhang, Y., Lucas, J. S., Dudko, O. K. & Murre, C. Chromosome dynamics near the sol-gel phase transition dictate the timing of remote genomic interactions. Nat. Commun. 10 , 2771 (2019).
Strickfaden, H. et al. Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cells. Cell 183 , 1772–1784 e1713 (2020).
Bagnoli, J. W. et al. Sensitive and powerful single-cell RNA sequencing using mcSCRB-seq. Nat. Commun. 9 , 2937 (2018).
Ziegenhain, C. et al. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65 , 631–643 e634 (2017).
Parekh, S., Ziegenhain, C., Vieth, B., Enard, W. & Hellmann, I. zUMIs—a fast and flexible pipeline to process RNA sequencing data with UMIs. Gigascience 7 , (2018).
Rau, A., Gallopin, M., Celeux, G. & Jaffrezic, F. Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 29 , 2146–2152 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339 , 823–826 (2013).
Link, S. et al. PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nat. Commun. 9 , 4300 (2018).
Punzeler, S. et al. Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation. EMBO J. 36 , 2263–2279 (2017).
Belaghzal, H., Dekker, J. & Gibcus, J. H. Hi-C 2.0: an optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123 , 56–65 (2017).
Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36 , 311–316 (2020).
Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9 , 999–1003 (2012).
Cremer, M. et al. Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods Mol. Biol. 463 , 205–239 (2008).
Kishi, J. Y. et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat. Methods 16 , 533–544 (2019).
Kishi, J. Y., Schaus, T. E., Gopalkrishnan, N., Xuan, F. & Yin, P. Programmable autonomous synthesis of single-stranded DNA. Nat. Chem. 10 , 155–164 (2018).
Bienko, M. et al. A versatile genome-scale PCR-based pipeline for high-definition DNA FISH. Nat. Methods 10 , 122–124 (2013).
Solovei, I. & Cremer, M. 3D-FISH on cultured cells combined with immunostaining. Methods Mol. Biol. 659 , 117–126 (2010).
Solovei, I. et al. in FISH: A Practical Approach (eds Beatty, B. et al.) 119–157 (Oxford Univ. Press, 2002).
Solovei, I. Fluorescence in situ hybridization (FISH) on tissue cryosections. Methods Mol. Biol. 659 , 71–82 (2010).
Eberhart, A. et al. Epigenetics of eu- and heterochromatin in inverted and conventional nuclei from mouse retina. Chromosome Res. 21 , 535–554 (2013).
Eberhart, A., Kimura, H., Leonhardt, H., Joffe, B. & Solovei, I. Reliable detection of epigenetic histone marks and nuclear proteins in tissue cryosections. Chromosome Res. 20 , 849–858 (2012).
Solovei, I. et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152 , 584–598 (2013).
Walter, J. et al. Towards many colors in FISH on 3D-preserved interphase nuclei. Cytogenet Genome Res. 114 , 367–378 (2006).
Bystricky, K., Heun, P., Gehlen, L., Langowski, J. & Gasser, S. M. Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. Proc. Natl Acad. Sci. USA 101 , 16495–16500 (2004).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing chromosome conformation. Science 295 , 1306–1311 (2002).
Rippe, K. Making contacts on a nucleic acid polymer. Trends Biochem. Sci 26 , 733–740 (2001).
Ou, H. D. et al. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357 , (2017).
Hajjoul, H. et al. High-throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res. 23 , 1829–1838 (2013).
Lucas, J. S., Zhang, Y., Dudko, O. K. & Murre, C. 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell 158 , 339–352 (2014).
Nuebler, J., Fudenberg, G., Imakaev, M., Abdennur, N. & Mirny, L. A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl Acad. Sci. USA 115 , E6697–E6706 (2018).
Download references
Acknowledgements
We are grateful to D. Hörl and J. Ryan for the help with ImageJ plugins and programming. We thank A. Maiser and K. Brandstetter for the help with high-resolution microscopy. We acknowledge J. Bates, D. Eick, P. Becker, M. Carmo-Fonseca, A. Olins and D. Olins and H.C.Macgregor for fruitful and insightful discussions. We thank D. Kralev and T. Suzuki for the help with animation. This work has been supported by the Deutsche Forschungsgemeinschaft grants (SO1054/1 and SP2202/SO1054/2 to I.S., SPP 2202/LE721/17-1 to H.L. and SFB1064 to I.S. and H.L.) and National Institutes of Health grants (HG007743 to H.L., HG003143 to J.D. and GM114190 to L.M. by the Center for 3D Structure and Physics of the Genome of NIH 4DN Consortium, DK107980). J.D. is an investigator of the Howard Hughes Medical Institute. I.S. is deeply thankful to H. C. Macgregor for his guidance.
Author information
Katharina Thanisch
Present address: Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach an der Riss, Germany
These authors contributed equally: Johannes Ribisel, Simon Ullrich.
Authors and Affiliations
Department of Biology II, Biozentrum, Ludwig-Maximilians University Munich (LMU), Planegg-Martinsried, Germany
Susanne Leidescher, Simon Ullrich, Yana Feodorova, Sebastian Bultmann, Katharina Thanisch, Christopher Mulholland, Heinrich Leonhardt & Irina Solovei
Institute for Medical Engineering and Science, and Department of Physics, Massachusetts Institute of Technology, Cambridge, MA, USA
Johannes Ribisel & Leonid Mirny
Department of Medical Biology, Medical University of Plovdiv; Division of Molecular and Regenerative Medicine, Research Institute at Medical University of Plovdiv, Plovdiv, Bulgaria
Yana Feodorova
Program in Systems Biology, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, MA, USA
Erica Hildebrand & Job Dekker
Skolkovo Institute of Science and Technology, Skolkovo, Russia
Alexandra Galitsyna
BioMedizinisches Center, Ludwig-Maximilians University Munich, Planegg-Martinsried, Germany
Stephanie Link
Howard Hughes Medical Institute, Chevy Chase, MD, USA
You can also search for this author in PubMed Google Scholar
Contributions
I.S. conceived the project. S. Leidescher, S.U., Y.F., K.T. and I.S. obtained biological samples. I.S., Y.F., S. Leidescher and S.U. conceived and performed microscopy and image analysis. S. Leidescher, C.M. and S. Link performed RNA-seq and ChIP–seq experiments. S.B. performed RNA-seq and ChIP–seq analyses. Y.F. and E.H. performed Hi-C experiments. E.H., J.D., J.R. (formerly known as J. Nübler), A.G. and L.M. performed Hi-C analysis. J.R. with contribution from L.M. performed simulations. S. Leidescher, S.U., J.R., A.G. and I.S. prepared the figures. I.S. wrote the manuscript with contributions from S. Leidescher, Y.F., S.U., J.R., K.T., E.H., H.L., J.D. and L.M.
Corresponding authors
Correspondence to Leonid Mirny or Irina Solovei .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Peer review
Peer review information.
Nature Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
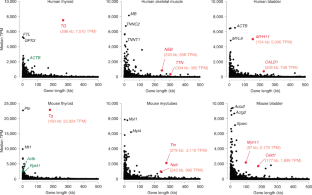
Extended data fig. 1 long genes are rare and expressed at lower levels than short genes..
a , Analysis of gene length distribution within the human and mouse genomes showed that about 43% and 46% of all protein coding genes, respectively, have a length ≤20 kb and only 18% and 14% have a length of 100 kb or above. Bin size: 20 kb. Genes are annotated according to GENCODE. Only genes with a length <500 kb are shown. b , To select suitable genes for visualization with light microscopy, we studied gene expression profiles across 50 human tissues using the publicly available Genotype-Tissue Expression database (GTEx Consortium) and found that long genes, as a rule, are not highly expressed. For example, in liver ( top ) and brain ( bottom ) there were no expressed genes with both a length ≥100 kb and with a median expression ≥1,000 TPM. c , Comparison of RNAPII occupancy between short and long expressed genes. ChIP-seq with an antibody against the CTD of RNAPII in cultured mouse myoblasts ( left ) and in vitro differentiated myotubes ( right ). All genes, expressed (>1 TPM, blue ) and silent (<1 TPM, red ), were split into five categories according to their size. RNAPII density ( Y-axis ) is plotted against the respective position within the gene ( X-axis ); each gene is divided into 200 equally sized bins and genes from the same size category are aligned according to the bins. Expressed genes display a higher occupancy with RNAPII compared to non-expressed genes, especially in the TSS region. In the group of expressed genes, the RNAPII occupancy negatively correlates with gene length: the shorter the genes, the higher the RNAPII occupancy. d , Analysis of RNA-seq data for myoblasts ( left ) and myotubes ( right ). The median expression level (TPM) is higher in groups containing shorter genes (<25 kb) and generally negatively correlates with gene length.
Extended Data Fig. 2 Visualization of the five selected genes in expressing and not expressing cells.
a , The Tg gene is expressed in thyrocytes where both alleles form prominent TLs expanding into the nuclear interior. In neighboring cells with a silent Tg gene - parathyroid gland cells, tracheal chondrocytes, epithelial cells, fibroblasts and muscles - Tg is highly condensed and sequestered to the nuclear periphery. b , The Ttn gene is expressed in skeletal muscle (b1), heart muscle (b2) and myotubes differentiated from Pmi28 myoblasts in vitro (b3). Note that only muscle nuclei ( solid arrowheads ) exhibit TLs. In muscle fibroblasts ( arrows ) or undifferentiated cultured myoblasts ( empty arrowheads ), Ttn is condensed at the nuclear periphery. c , The Neb gene is expressed in skeletal muscles and cultured myotubes, although to a lesser degree than Ttn . Accordingly, it forms smaller TLs. Arrowheads indicate muscle nuclei; arrows indicate fibroblast nuclei with silent Neb . d, e , The Myh11 (d) and Cald1 (e) genes are expressed in smooth muscles of colon and bladder where they form TLs. Note that after RNA-FISH, only smooth muscles ( arrowheads ) but not the neighboring epithelial cells ( arrows ) exhibit TLs. In addition, Cald1 is expressed in cultured myoblasts and forms small TLs in these cells. As indicated above the panels, images display signals after either RNA-FISH (no tissue/cell DNA denaturation and no RNasing), or simultaneous detection of DNA and RNA (tissue/cell DNA denaturation but no RNasing). All images are projections of 1–3 µm confocal stacks. Scale bars for overviews of skeletal muscle, colon and bladder, 50 µm ; for the rest of the panels, 5 µm . Data represent 100 in a,b3 and 10 in b1,b2,c-e independent experiments.
Extended Data Fig. 3 Structure and compaction of TLs.
a , The internal structure of Tg and Ttn TLs is not resolvable after deconvolution ( left ) and high resolution microscopy ( mid, right ). b , Coiling and folding of TLs demonstrated in 50–70 nm thin resin sections. The upper panel shows thin sections through nuclei of thyrocytes stained with DAPI ( red ) and Tg TLs detected by RNA-FISH ( green ). The lower panel shows 2-fold close-ups of the corresponding Tg TL as grey-scale images. Note curling and twisting of the loops. Images are single optical sections. c , To assess the compaction level of TLs, the contour length of three Tg TL regions was measured on projections after RNA-FISH. The track of the Segmented Line tool in ImageJ , used for measurements, is shown on the right panel . Tg regions of 153 kb, 109 kb and 62 kb had a similar compaction level and measured 9 µm, 6 µm and 4 µm, respectively. These values correspond to a nucleosomal structure of chromatin ( table on the right ). However, since Tg TLs display internal structures and since the measurements were performed on maximum intensity projections, the compaction level of Tg TLs is probably overestimated. Scale bars: a, 2 µm ; b,c, 1 µm . Data represent 3 independent experiments in a-c.
Extended Data Fig. 4 TLs manifest co-transcriptional splicing.
Two sequentially positioned introns were labeled with oligoprobes encompassing 1.2–5 kb. The schematics above the panels depict the distribution of oligoprobes ( green and red rectangles ), labeled introns ( green and red lines ) and positions of BAC probes used as references ( grey lines above genes ). After RNA-FISH the intron probes label TLs only partially and sequentially. Since the 5’ and 3’ intron signals do not overlap, the 5’ introns are spliced before the 3’ introns are read. For instance, in Cald1 , introns 1 and 3 are separated by intron 2, suggesting that the “green” intron 1 is spliced out before polymerases reach the “red” intron 3, most likely after RNAPII runs over the 3’ splice-site of the first intron. Projections of confocal sections through 2–3.5 µm . Scale bars: 2 µm , in close-ups, 1 µm . Data represent 2 independent experiments.
Extended Data Fig. 5 Close association of TLs with splicing factors.
Splicing factors (SON) and components of exon-junction complexes EIF4A3 and RBM8A (Y14), are either co-stained with RNAPII Ser2P ( the two top rows ) or visualized together with TLs in immuno-FISH ( the rest of the rows ). Note that signals of TLs and splicing factors colocalize only partly. The myotube nucleus is tetraploid and thus exhibits 4 Ttn RNA signals. Images are partial projections of either 0.6 µm (for immunostaining) or 0.9 µm (for immuno-FISH). Scale bars: 2 µm , in close-ups, 1 µm . Data represent 3 independent experiments.
Extended Data Fig. 6 Nucleoplasmic granules in cells with highly expressed long genes.
a , RNA-FISH reveals numerous nucleoplasmic granules ( arrows ) surrounding TLs ( arrowheads ) after hybridization with genomic probes. For clarity, only RNA signals are shown within the outlined nuclei. Empty arrowheads point at similar granules in the myotube cytoplasm. The asterisk marks the nucleus of a myoblast not expressing Ttn . b , In muscles and cultured myotubes, the majority of granules (81%) are double-labeled with probes for the 5’ and 3’ halves of Ttn and found in both the nucleoplasm and cytoplasm ( arrows on the lower panel ), thus likely representing Ttn mRNAs. Remarkably, the 5’ and 3’ signals are spatially distinguished within the granules ( insertion ) presumably due to the exceptionally long Ttn mRNA of ca 102 kb. The observed separation of the 5’ and 3’ halves of Ttn mRNA is in agreement with previously described structures of cytoplasmic mRNPs 28 , 29 , 30 . c , In difference to Ttn , mRNAs of Tg, Neb, Cald1 and Myh11 genes are short (4–20 kb) and can be only detected with oligoprobes specifically hybridizing to all exons. Thus the oligoprobe for all 48 Tg exons hybridizes to nRNAs decorating TLs ( arrows ) and also labels multiple nucleoplasmic granules ( arrowheads ). d , The majority of the other thyrocyte nucleoplasmic granules are labeled with either 5’ ( green ) or 3’ ( red ) genomic probes with only 10% of granules being double-labeled. The brightness of the RNA-signal on the most right panel is purposely increased to highlight nucleoplasmic granules ( green and red arrows ). Such differential labeling of nucleoplasmic granules, exemplified here for thyrocytes, is characteristic for other studied genes and strongly suggests that these granules represent accumulations of excised introns. The distribution of the used BAC probes in respect to the studied genes are depicted above the image panels. Scale bars: a, 2 µm for Tg and Myh11 , 5 µm for Ttn and Neb ; b, c, d, 2 µm . Data represent 10 in a,b and 3 in c,d independent experiments.
Extended Data Fig. 7 Fragments of Hi-C maps around TL-forming genes in corresponding tissues and cells.
Each row shows the same region around indicated genes in different tissues or cells; “on” indicates a cell type where a gene is active, allowing to compare changes associated with TL formation. Blue arrows indicate loss of TAD borders and associated dots of CTCF-CTCF enrichment for expressed Tg and Myh11 . Red arrows indicate an increase in self-interactions within genes visible for expressed Ttn, Neb and Myh11 .
Extended Data Fig. 8 Cis-to-trans contact ratios and A/B compartments in studied cells.
a , Cis-to-trans ratios and compartment affiliations for 5 studied genes. The scatter plots are computed from the compartment profiles and the cis-to-trans ratio profiles of the chromosomes harboring the genes at a bin size of 32 kb. Genes of interest are highlighted with red dots; the white crosses mark the chromosome means of the compartment and cis-to-trans ratio profiles. Tg, Ttn, Neb and Myh11 move from B to A compartment upon their activation; Cald1 is found in A compartment not only in myoblasts and smooth muscle, as expected, but also in thyroid and myotubes. This is in agreement with the low Cald1 expression also in thyroid samples enriched in blood vessels and in samples of myotubes that normally include up to 20% of myoblasts, as well as with localization of Cald1 in a gene-dense region of chromosome 6. Note that the Ttn and Neb genes tend to be in A compartment, although to a lesser degree, also in myoblasts, which can be explained by the presence of cells that started their differentiation into single-cell myotubes. b , Cis-to-trans ratios are lower in A than in B compartments for all four studied cell types. The scatter plots are computed from the genome wide compartment profile and the cis-to-trans ratio profile, both at a bin size of 1,024 kb. The Pearson correlation coefficients are indicated in the upper right corners of the scatter plots. c , Externalization of the expressed genes from their harboring chromosomes measured by the cis-to-trans ratios as a function of gene length and expression in corresponding tissues. Left column of heatmaps : median cis-to-trans normalized by chromosome. Notice the reduction of cis-to-trans (that is, increasing externalization) for highly transcribed genes (top rows in each heatmap) as gene length increases (moving from left to right). Similarly, cis-to-trans goes down for long genes ( right-most column of each heatmap) as expression increases (going up along this column). Interestingly, lowly transcribed long genes ( lower right corner ) have high cis-to-trans , indicating strong internalization, but become strongly externalized as they become highly expressed ( upper right corner ). Middle column : median cis-to-trans ratios controlled for compartmental signal (the first eigenvector, E1). Heatmaps show the logarithm of observed median cis-to-trans ratio divided by the expected given E1 in the corresponding bin. For the expected value, all the genomic bins were separated into 20 ranges by their E1, and the median cis-to-trans for each range was considered as expected. Notice that for most genes, E1 explains most of the cis-to-trans ratio. However, cis-to-trans is considerably lower for extremely long and extremely highly transcribed genes ( upper right corner of each heatmap). Right column : the number of genomic bins in each range of length and transcription. Notice that very few genes show high externalization. d , Table of coefficients of determination (R2) for regression of cis-to-trans ratios of the genomic bins normalized by chromosome, in four tissues. Only the bins of expressed genes (TPM > 100) are considered. Gene length is an excellent predictor of cis-to-trans ratio for genomic bins of highly expressed genes (TPM > 1000, 1st row ), but not for other expressed genes (TPM from 100 to 1000, 2nd row ). Gene expression is a good predictor of cis-to-trans ratio for genomic bins of long genes (TPM > 50 kb, 3rd row), but not for shorter genes (TPM < 50 kb, last row). Heatmaps : visual illustration of gene subsets in this analysis.
Extended Data Fig. 9 TLs do not cause insulation at different length scales.
Insulation assesses Hi-C contacts spanning across a given locus up to a maximal distance w ( top right insert ). Contacts in a square window of size w were aggregated and the square was slid along the Hi-C diagonal. The score was normalized by its genome wide mean. Profiles show log2 of the score, such that a locus with profile value -1 has a two-fold reduced number of contacts spanning the locus up to distance w compared to the genome wide mean. Insulation scores are computed with the cooltools package ( https://github.com/mirnylab/cooltools ). We computed insulation profiles for Hi-C maps with a bin size of 128 kb for various window sizes from 256 kb up to ≈16 Mb. For every analyzed gene, the left and right columns show a 3 and 20 Mb Hi-C map with insulation profiles for different window sizes; the top and bottom panels show insulation profiles in expressing ( on ) and non-expressing ( off ) cells, respectively. The analysis shows little correlation between insulation and the formation of TLs: insulation profiles at the gene loci do not differ much between cell types with the gene on or off . For example, the Tg gene shows a moderate dip at scales up to ≈ 1 Mb in both thyroids ( on ) and myoblasts ( off ), and no dip in either cell type on the larger scale. Analysis of simulated TLs (Fig. 8 and Extended data Fig. 10 ) confirmed that TL formation does not cause large scale insulation ( bottom row ).
Extended Data Fig. 10 Polymer simulation of chromosomes.
a , Six chromosomes (50 Mb each) were initiated in a mitotic-like state with unit volume density. Row 1 and 3 show top views, row 2 and 4 show side views. In rows 1 and 2 six chromosomes are differentially colored; in rows 3 and 4 compartmental segments of A and B type chromatin are differentially colored with red for A and blue for B compartments. The initial expansion is very fast (column 2). However, once the chromosomes fill the nucleus uniformly, the subsequent dynamics is very slow and chromosomes retain their territoriality (note that times increase logarithmically). Nevertheless, due to attraction of B-type chromatin to the lamina, a radial structure emerges (rows 3 and 4). b , TLs are modeled by choosing a 300 kb segment on each chromosome 25.4 minutes after expansion and increasing the stiffness of the polymer fiber. The genes quickly expand on the order of minutes and are simulated for approximately 1.5 h. The measurements of inter-flank distances and Hi-C maps are performed using configurations sampled from the second half of this time interval. When genes are deactivated by removing the excess stiffness, they collapse back to the inactive state. c , Left: Hi-C of all 6 chromosomes shows their territoriality as patches. Second-left: A Hi-C contact map averaged over all 6 chromosomes exhibits the checkerboard pattern of a typical segregation of A- and B-type chromatin. The three rightmost graphs show zoomed views of modeled genes with stiffness profiles above the maps.
Supplementary information
Supplementary information.
Supplementary Figs. 1–5.
Reporting Summary
Peer review information, supplementary table 1.
Excel spreadsheet includes the distribution of human genes according to their lengths, a list of protein-coding genes and their expression levels in different human tissues, and a list of selected genes with lengths larger than 100 kb and expression levels of ca. 1,000 TPM.
Supplementary Table 2
Excel spreadsheet includes the distribution of mouse genes according to their lengths, a list of protein-coding genes and their expression levels in two mouse tissues and two cultured cell types, a list of five selected genes with lengths of ca. 100 kb and expression levels of ca. 1,000 TPM and a list of long lowly expressed genes used for comparison of cis -to- trans contact ratios with the studied genes.
Supplementary Table 3
Excel spreadsheet includes (1) the list and schematics of genomic BAC probes used for DNA and RNA FISH experiments; (2) primer pairs used for introduction of protospacer sequences into U6-gRNA-GFP-H2A; (3) primer sequences used to verify the expression of full length Ttn after activation with dCas9-VPR and the corresponding gel image; (4) primer pairs used to amplify Tg cDNA regions containing exons 2–12 and 33–47; (5) primers for generation of oligoprobes for Tg intron 41 (HD FISH); (6) primers for generation of oligoprobes for Tg 5′ exons and introns (SABER FISH); (7) primers for generation of probes for all Tg exons, intron 41 and the Sla gene; (8) primers for generation of probes for Ttn and Cald1 introns (SABER FISH).
Supplementary Table 4
Excel spreadsheet includes (1) the total number of acquired nuclei from five tissues and two cultured cell types, as well as the number of nuclei with a single expressed allele; (2) information on the number of reads in Hi-C experiments.
Supplementary Video 1
Confocal stacks through nuclei of mouse thyrocytes (counterstained with DAPI, red) after RNA FISH with a genomic probe for the Tg gene (green). Note the volatile shape of the Tg TLs and their great expansion into the nuclear interior.
Supplementary Video 2
Cartoon showing how transcription initiation and termination of a highly expressed gene lead to formation and disappearance of a TL. The sequence of events: a gene body (orange thread) is coiled within a locus (blue threads) in a compact structure; upon transcription initiation, RNAPIIs (dark-grey oval structures) are loading at the gene promoter (red) and begin to elongate; during elongation, nRNPs appear and grow in size (depicted as grey amorphous structures); during a transcription pause, chromatin of the gene is coiled and forms a sliding knot (orange thread), dynamically formed beyond the last RNAPII of the first burst and disentangled by first RNAPII of the second burst; dense decoration with voluminous nRNP rigidifies the gene axis and forces its expansion, as well as the divergence of gene flanks (blue threads); RNAPIIs of the first transcription burst reach the 3′ gene end, release attached nRNPs and dissociate from the gene; the sliding chromatin knot reaches the 3′ gene end; RNAPIIs stop loading at the promoter at the end of the second burst; gene condensation starts 5′-terminally; the gene flanks begin to converge; the gene returns to its coiled compact state.
Source data
Source data fig. 3.
Statistical source data.
Source Data Fig. 5
Source data fig. 7, rights and permissions.
Reprints and permissions
About this article
Cite this article.
Leidescher, S., Ribisel, J., Ullrich, S. et al. Spatial organization of transcribed eukaryotic genes. Nat Cell Biol 24 , 327–339 (2022). https://doi.org/10.1038/s41556-022-00847-6
Download citation
Received : 09 April 2021
Accepted : 10 January 2022
Published : 17 February 2022
Issue Date : March 2022
DOI : https://doi.org/10.1038/s41556-022-00847-6
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Co-transcriptional gene regulation in eukaryotes and prokaryotes.
- Morgan Shine
- Jackson Gordon
- Karla M. Neugebauer
Nature Reviews Molecular Cell Biology (2024)
Transcriptional condensates: a blessing or a curse for gene regulation?
- Martin Stortz
- Diego M. Presman
- Valeria Levi
Communications Biology (2024)
Chromatin alternates between A and B compartments at kilobase scale for subgenic organization
- Hannah L. Harris
- M. Jordan Rowley
Nature Communications (2023)
Transcripts in the loop
- Eytan Zlotorynski
Nature Reviews Molecular Cell Biology (2022)
The sight of transcription
- Elias T. Friman
- Wendy A. Bickmore
Nature Cell Biology (2022)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

Regulation of gene expression and the transcription factor cycle hypothesis
Affiliation.
- 1 Inst. J. Monod, CNRS and University Paris Diderot, 9, rue Larrey, 75005 Paris, France.
- PMID: 22234303
- DOI: 10.1016/j.biochi.2011.12.010
Post-genomic data show unexpected extent of the transcribed genome and the size of individual primary transcripts. Hence, most cis-regulatory modules (CRMs) binding transcription factors (TFs) at promotor, enhancer and other sites are actually transcribed within full domain transcripts (FDTs). The ensemble of these CRMs placed way upstream of exon clusters, downstream and in intronic or intergenic positions represent a program of gene expression which has been formally analysed within the Gene and Genon concept [1,2]. This concept has emphasised the necessity to separate product information from regulative information to allow information-theoretic analysis of gene expression. Classically, TFs have been assumed to act at DNA level exclusively but evidence has accumulated indicating eventual post-transcriptional functions. The transcription factor cycle (TFC) hypothesis suggests the transfer of DNA-bound factors to nascent RNA. Exerting downstream functions in RNA processing and transport, these factors would be liberated by RNA processing and cycle back to the DNA maintaining active transcription. Sequestered on RNA in absence of processing they would constitute a negative feedback loop. The TFC concept may explain epigenetic regulation in mitosis and meiosis. In mitosis control factors may survive as single proteins but also attached to FDTs as organised complexes. This process might perpetuate in cell division conditioning of chromatin for transcription. As observed on lampbrush chromosomes formed in avian and amphibian oogenesis, in meiosis the genome is fully transcribed and oocytes conserve high Mr RNA of high sequence complexity. When new interphase chromosomes form in daughter cells and early embryogenesis, TFs and other factors attached to RNA might be reinserted onto the DNA.
Copyright © 2011 Elsevier Masson SAS. All rights reserved.
- Chromosomes / genetics
- Chromosomes / metabolism
- Gene Expression Regulation*
- Genes, Viral
- Models, Genetic
- Protein Binding
- RNA Processing, Post-Transcriptional
- RNA, Messenger / genetics
- RNA, Messenger / metabolism*
- Regulatory Sequences, Ribonucleic Acid
- Transcription Factors / metabolism*
- Transcription, Genetic
- RNA, Messenger
- Transcription Factors
- Research Article
- Chromosomes and Gene Expression
Kinetic competition during the transcription cycle results in stochastic RNA processing
- Antoine Coulon
- Matthew L Ferguson
- Valeria de Turris
- Murali Palangat
- Carson C Chow
- National Institutes of Health, United States ;
- National Cancer Institute, National Institutes of Health, United States ;
- Istituto Italiano di Tecnologia, Italy ;
- Open access
- Copyright information
Share this article
Cite this article.
- Daniel R Larson
- Copy to clipboard
- Download BibTeX
- Download .RIS
eLife digest
Introduction, materials and methods, article and author information.
Synthesis of mRNA in eukaryotes involves the coordinated action of many enzymatic processes, including initiation, elongation, splicing, and cleavage. Kinetic competition between these processes has been proposed to determine RNA fate, yet such coupling has never been observed in vivo on single transcripts. In this study, we use dual-color single-molecule RNA imaging in living human cells to construct a complete kinetic profile of transcription and splicing of the β-globin gene. We find that kinetic competition results in multiple competing pathways for pre-mRNA splicing. Splicing of the terminal intron occurs stochastically both before and after transcript release, indicating there is not a strict quality control checkpoint. The majority of pre-mRNAs are spliced after release, while diffusing away from the site of transcription. A single missense point mutation (S34F) in the essential splicing factor U2AF1 which occurs in human cancers perturbs this kinetic balance and defers splicing to occur entirely post-release.
To make a protein, part of a DNA sequence is copied to make a messenger RNA (or mRNA) molecule in a process known as transcription. The enzyme that builds an mRNA molecule first binds to a start point on a DNA strand, and then uses the DNA sequence to build a ‘pre-mRNA’ molecule until a stop signal is reached.
To make the final mRNA molecule, sections called introns are removed from the pre-mRNA molecules, and the parts left behind—known as exons—are then joined together. This process is called splicing. However, it is not fully understood how the splicing process is coordinated with the other stages of transcription. For example, does splicing occur after the pre-mRNA molecule is completed or while it is still being built? And what controls the order in which these processes occur?
One theory about how the different mRNA-making processes are coordinated is called kinetic competition. This theory states that the fastest process is the most likely to occur, even if the other processes use less energy and so might be expected to be preferred. Alternatively, the different steps may be started and stopped by ‘checkpoints’ that cause the different processes to follow on from each other in a set order.
Coulon et al. used fluorescence microscopy to investigate how mRNA molecules are made during the transcription of a human gene that makes a hemoglobin protein. To make the RNA visible, two different fluorescent markers were introduced into the pre-mRNA that cause different regions of the mRNA to glow in different colors. Coulon et al. made the introns fluoresce red and the exons glow green. Unspliced pre-mRNA molecules contain both introns and exons and so fluoresce in both colors, whereas spliced mRNA molecules contain only exons and so only glow with a green color.
By looking at both the red and green fluorescence signals at the same time, Coulon et al. could see when an intron was spliced out of the pre-mRNA. This revealed that in normal cells, splicing can occur either before or after the RNA is released from where it is transcribed. Thus, splicing and transcription does not follow a set pattern, suggesting that checkpoints do not control the sequence of events. Instead, the fact that a spliced mRNA molecule can be formed in different ways suggests kinetic competition controls the process.
In some cancer cells, there are defects in the cellular machinery that controls splicing. When looking at cells with such a defect, Coulon et al. found that splicing only occurred after transcription was completed. This study thus provides insight into the complex workings of mRNA synthesis and establishes a blueprint for understanding how splicing is impaired in diseases such as cancer.
Co-transcriptional processing of nascent pre-mRNA is a central mechanism for gene regulation in eukaryotes and requires temporal coordination between transcription initiation, elongation, splicing, and cleavage. Each of these processes is carried out by megadalton macromolecular complexes acting at a single genetic locus, and kinetic competition between these processes has been proposed to determine RNA fate ( Bentley, 2014 ). Genome-wide studies across organisms indicate heterogeneous distributions of both RNA polymerase and nascent RNA along the gene, suggesting that kinetic checkpoints exist throughout the gene, including at promoter-proximal sites, translation start sites, intron–exon boundaries, and at the 3′ end of genes ( Core et al., 2008 ; Nechaev et al., 2010 ; Churchman and Weissman, 2011 ; Hah et al., 2011 ; Larson et al., 2014 ). However, population studies reflect the balance of kinetic rates and are unable to resolve the multiple competing processes occurring at a single gene. Moreover, genome-wide measurements lack the time-resolution which might provide mechanistic clues about the underlying enzymatic processes.
The hypothesis of kinetic competition is that a fast process will out-compete a process which may in fact be more energetically preferred. Kinetic competition during the transcription cycle has been shown to influence splice site selection during alternative splicing ( de la Mata et al., 2003 ), recruitment of factors which release promoter-proximal pausing ( Li et al., 2013 ), and even RNAi-mediated genome defense ( Dumesic et al., 2013 ). Since these processes occur within the dynamic milieu of the nucleus, the stochastic interactions between macromolecules may result in a range of possible outcomes for the nascent RNA. Stochastic RNA synthesis—the phenomenon whereby the inherently stochastic nature of bio-molecular encounters and reactions leads to a non-deterministic production of transcripts—has been directly visualized in multiple organisms ( Golding et al., 2005 ; Chubb et al., 2006 ; Yunger et al., 2010 ; Larson et al., 2011 , 2013 ). Yet, stochastic RNA processing—the possibility that stochastic bio-molecular reactions might lead to non-deterministic pathways/outcomes in the making and maturation of an RNA—has never been directly observed, and the potential consequences for gene regulation are largely unexplored. Alternatively, regulatory checkpoints have been proposed which safeguard against such stochastic RNA processing events, providing a level of quality control. For example, the model of exon definition requires that splicing of the terminal intron relies on synergy between 3′ end formation, nascent RNA cleavage, and intron excision ( Berget et al., 1977 ; Niwa et al., 1990 ). Similarly, multiple studies indicate an increased density of nascent RNA present at the 3′ end of genes or in the chromatin-bound fraction, suggesting that nascent RNA is retained at the site of transcription to ensure correct processing ( Glover-Cutter et al., 2008 ; Brody et al., 2011 ; Carrillo Oesterreich et al., 2010 ; Bhatt et al., 2012 ). In both the competition model and the checkpoint model, kinetics plays a prominent role, but in the latter case, the cell has developed additional safeguard mechanisms.
In this article, we use an in vivo single-molecule RNA imaging approach to directly measure kinetic coupling between transcription and splicing of a human β-globin reporter gene. The approach is based on simultaneous dual-color imaging of both the intron and exon of the same pre-mRNA using both PP7 and MS2 stem loops ( Bertrand et al., 1998 ; Chao et al., 2008 ). We find that kinetic competition results in multiple competing pathways for pre-mRNA splicing. Splicing of the terminal intron occurs stochastically both before and after transcript release, indicating there is not a strict quality control checkpoint. Post-release splicing occurs from freely diffusing transcripts in the nucleus and is an order of magnitude faster than splicing at the site of transcription. A single missense mutation (Ser34Phe) in the zinc finger domain of the conserved splicing factor U2AF1 which is recurrent in multiple cancers ( Yoshida et al., 2011 ; Graubert et al., 2012 ; Waterfall et al., 2014 ) changes the balance, making all splicing post-release. This same effect can also be observed on the endogenous, un-modified fragile X mental retardation mRNA ( FXR1 ). Our results show that kinetic competition governs the stochastic balance between multiple competing pathways for RNA synthesis and processing and that this balance is perturbed by oncogenic mutations.
Real-time visualization of transcription and splicing at the single-molecule level in living cells
To visualize transcription, splicing, and release of single transcripts in living cells, we used time-lapse fluorescence microscopy of multiply labeled RNA ( Bertrand et al., 1998 ). We stably integrated into U2-OS cells, a human β-globin reporter with a DNA cassette that encodes for 24X PP7 RNA hairpins in the second intron ( Chao et al., 2008 ) and a 24X MS2 hairpin cassette in the 3′ UTR ( Boireau et al., 2007 ) ( Figure 1A ). The constitutively expressed PP7-coat protein tagged with mCherry (PCP-mCherry) and MS2-coat protein tagged with GFP (MCP-GFP) bind with high affinity (the on rate for MCP is 0.54 μM –1 s –1 . At a nuclear concentration of 1 μM, the average time for the MCP to bind a completed stem loop is 1.85 s) ( Buenrostro et al., 2014 ) to the RNA stem loops as homodimers, tagging each cassette with 48 fluorophores of a single color, resulting in orthogonal labeling of two different parts of the nascent transcript ( Hocine et al., 2013 ; Martin et al., 2013 ; Buenrostro et al., 2014 ). Since the PP7 cassette is intronic, unspliced RNAs appear in both colors, while spliced RNAs are only visible in green. Time-lapse imaging of cells in 3D reveals a temporally fluctuating diffraction-limited spot, co-localizing in both colors, that corresponds to the transcription site (TS) where nascent transcripts are synthesized ( Figure 1B–C , Videos 1 and Videos 2 ). We observed mature mRNA (exon only) diffusing in both the nucleus and the cytoplasm ( Video 3 ), and we verified expression of the protein product, indicating the message is spliced and translated correctly ( Figure 1—figure supplement 1B–C ).

Real-time measurement of transcription and splicing in living cells.
( A ) Schematic of the human β-globin report gene construct. Reporter splicing efficiency >95% by qRT-PCR ( Figure 1—figure supplement 1C ). ( B ) 3D images of diffraction-limited spot in both channels corresponding to the transcription site (TS, arrow). Bar: 4 µm. ( C ) Fluorescence fluctuations recorded at the TS reflect stochastic transcriptional events. Dotted lines are background traces recorded in the nucleus, 8 µm away from the TS. ( D and E ) Examples of pre- and post-release splicing observed when the intron (red signal) disappears simultaneously with ( D ) or before ( E ) the exon (green signal).

This video cannot be played in place because your browser does support HTML5 video. You may still download the video for offline viewing.
Time-fluctuating transcription sites.
Cells show a diffraction-limited fluorescent spot colocalizing in both colors (red: intron, green: exon), corresponding to the transcription site of the reporter gene. The fluorescence intensity of each site fluctuates over time as nascent transcripts are synthesized, spliced, and released from the transcription site. Large orange shapes in nuclei are nucleoli (Ferguson and Larson, 2013 ).

Tracking of a transcription site in 4D.
The video shows, for the intron and exon signals (left and center panels), the maximum intensity projected image from the top (square image) and from the sides (rectangle images), revealing the transcription site (TS) in three dimensions (3D) and over time (4D). Image analysis is used to track the TS over time in both colors. The blue box and cross indicate the location of the TS as found by the tracking algorithm. The right panel is the merge of both signals.

Spliced RNAs diffusing in the nucleus and the cytoplasm.
Cells are imaged here with a high laser power and a short exposure time so that diffusion of single RNAs can be appreciated. It reveals a population of transcripts diffusing in both the nucleus and the cytoplasm, as evidenced by fast fluctuations observed in the exon signal (right panel). These transcripts are, for the most part, already spliced since the intron signal (center panel) does not show the same fluctuations. In these imaging conditions, unspliced transcripts are only visible at the transcription site (bright spot in the nucleus colocalizing in both color; see merge in left panel).
By simultaneously observing the fluorescence intensity of the intron and the exon of a single nascent transcript, one can determine when the intron is excised from the pre-mRNA. We find that both pre- and post-release splicing are visible as single events at the same gene over time ( Figure 1C ). In the case of post-release splicing, the intronic fluorescence appears, followed by the exon fluorescence, followed by a coincidental drop in both colors reflecting the release of an unspliced RNA ( Figure 1D and Figure 1—figure supplement 1E ). For pre-release splicing, there is a delay between the drop-off of the red and green signals, indicating intron removal before release ( Figure 1E ). Diffusion of the pre-mRNA away from the TS is rapid, which accounts for the precipitous drop of the signal in the time trace ( Figure 1C ).
Fluctuation analysis reveals the stochastic synthesis and processing kinetics of single transcripts
Most of the time, multiple nascent RNA is present at the TS, necessitating a general analysis method for extracting kinetic information from the time traces ( Larson et al., 2011 ). We developed a dual-color fluctuation correlation analysis approach for analyzing the complete transcription cycle, resulting in two temporal auto-correlation functions and a single temporal cross-correlation function ( Figure 2A ; See ‘Materials and methods’). These functions measure, over many traces, how a fluctuation in one fluorescence channel is statistically correlated with a fluctuation in either the same or the other channel after a given time delay.

Transcription and splicing kinetics are revealed by fluctuation analysis of dual-color fluorescence intensity time traces.
( A ) Auto- and cross-correlation functions quantify statistically correlated fluctuations occurring at different time delays, respectively within the same or between two signals. ( B ) Correlation functions ( G(τ) ) of experimental time traces (N = 21). Auto-correlations (red and green curves) are symmetrical by construction. Cross-correlations (blue and magenta curves) are two halves of a single continuous curve. Inset: short-delay behavior of the cross-correlation reveals that 13 ± 5% of the RNAs are spliced pre-release (p-value: pre-release fraction ≠ 0% and 100%; z-test). ( C ) Schematic representing stochastic pre- and post-release splicing. Purely pre-release splicing imposes the cross-correlation to have the same rising slope on both sides of the y-axis, while purely post-release makes the intron-to-exon cross-correlation (blue curve, positive delay) start as a plateau. The change of slope at τ = 0 delay is indicative of the fraction of splicing events occurring before release. ( D ) Spliceostatin A abolishes pre-release splicing. ( E ) Camptothecin delays the decay of the intron-to-exon cross-correlation and increases the pre-release fraction. All correlation functions are normalized by the value of the cross-correlation at 0 delay ( G rg (0) ). Error: SEM (bootstrap). Control correlation functions are shown in Figure 2—figure supplement 1G–H .
The correlation functions for β-globin ( Figure 2B , Figure 2—figure supplement 1A–B ) reveal the kinetic features hidden in the fluorescence time traces and encapsulate the stochastic kinetics of single-transcript synthesis ( Figure 2—figure supplement 2 , Supplementary file 1 —§2). At short time delays (<40 s), the cross-correlation reflects the order of splicing and release events ( Figure 2B–C and Supplementary file 1 —§2). If splicing never occurs before transcript release, the intron-to-exon cross-correlation (blue) starts horizontally at positive delays. On the other hand, if splicing always occurs before release, this function starts with a positive slope in alignment with its negative counterpart (magenta). In the case where splicing is stochastic and both outcomes may occur, their relative probability of occurrence is given by the change of slope of the cross-correlation at 0 delay ( Figure 2C , Figure 2—figure supplements 2D and 3 ). The experimental cross-correlation determined from ∼2000 individual β-globin transcripts indicates that splicing occurs before release for a fraction of transcripts (13 ± 5%, Figure 2B inset). We note that this change in slope at short time delay only denotes the relative order of splicing vs release but says nothing about the kinetics of these two processes. In summary, these data demonstrate that splicing and release are not firmly constrained to occur in a specific order (p < 0.003).
At longer delays (>40 s), other features of the transcription cycle are visible. For example, the delay at which the intron-to-exon cross-correlation ( Figure 2B , blue circles) starts decreasing (∼60 s) corresponds to the elongation time between the two cassettes (2573 bases apart), resulting in an elongation rate of ∼2.6 kb/min. Finally, the decay at time-scales > 100 s relates to the dwell time of transcripts at the TS ( Figure 2—figure supplement 2A,B ). Specifically, the long decays observed in all correlation functions indicates that RNA is not immediately released after transcription of the poly(A) site. Rather, the transcript remains at the TS for a duration which reflects either a pause at/near the poly(A) site, transcription past the termination site, or a post-cleavage retention of the transcript within the diffraction-limited spot ( Hofer and Darnell, 1981 ; Glover-Cutter et al., 2008 ; Brody et al., 2011 ; Carrillo Oesterreich et al., 2010 ; Bhatt et al., 2012 ).
To confirm our assignment of transcription cycle events to features in the correlation curve, we treated cells with drugs known to affect different aspects of RNA synthesis. The splicing inhibitor spliceostatin A (SSA) ( Kaida et al., 2007 ) abolished splicing at the TS as evidenced by the disappearance of the rise in the intron-to-exon correlation function ( Figure 2D ). Treatment with camptothecin (CPT, a topoisomerase I inhibitor known to slow down elongation [ Singh and Padgett, 2009 ]) resulted in a marked shift of the decreasing part of the intron-to-exon cross-correlation to longer delays ( Figure 2E ), which is the expected manifestation of slower elongation ( Figure 2—figure supplement 2A–B , Supplementary file 1 —§2).
As an additional control, when shuffling channels between traces or when using time traces recorded away from the TS, the correlation functions are flat ( Figure 2—figure supplement 1G–H ), supporting the fact that the correlation functions shown on Figure 2B reflect the molecular events happening at the TS.
Finally, we emphasize that an essential advantage of this approach is that correlation functions reveal single-transcript kinetics even from signals where multiple transcripts are present at any given time ( Supplementary file 1 —§1). As illustrated in Figure 3 , a single transcription and splicing event results in correlation functions with a peak near zero delay, reflecting the intra -transcript kinetics ( Figure 3A ). If three transcripts are present, additional peaks appear at non-zero delay, due to inter -transcript kinetics but all the correlations resulting from intra -transcript kinetics accumulate around 0 ( Figure 3B ). After averaging over many transcription and splicing events, inter -transcript correlations disappear, leaving only a central peak which reflects the kinetics of single transcripts ( Figure 3C ). See also Video 4 and Figure 3—figure supplement 1 .

Correlation functions reflect single-transcript kinetics.
( A ) A dual-color time trace with a single transcription event yields correlation functions with features around 0 delay and flat elsewhere. ( B ) When several transcription events are present in a time trace, the correlation coming from each individual RNA accumulates around 0 delay, while all the correlation between pairs of RNAs distributes uniformly on the delay axis. ( C ) When there are many transcription events per time trace and/or many traces are used to produce an average correlation function, the correlation from single transcripts dominates and that from pairs of transcripts averages out. The resulting correlation functions hence reflect single-transcript kinetics. Time traces shown are simulations where the statistics of transcript kinetics are similar to those we measured by live cell imaging. Traces in ( C ) have the same duration and number of transcripts as estimated in experimental data (e.g., Figure 1C ). See Video 4 for an animation of how the correlation functions converge as the number of transcripts increases.

Correlation functions reveal single transcript kinetics.
This video shows the convergence of the correlation functions for increasing number of transcripts in a time trace. See also Figure 3 .
Kinetic competition between splicing and transcript retention determines the balance between pre- and post-release splicing
The preceding conclusions are general and make no reference to a specific model. To gain further insight, we developed mathematical models which relate the shape of the correlation functions to the timing of the underlying molecular processes (see ‘Materials and methods’). We generated five different mechanistic schemes: (I) purely post-release splicing, (II) independence between splicing and elongation/release, (III) polymerase pausing at the 3′ splice site (ss) until splicing is complete, (IV) splicing only during 3′ end retention of the transcript, and (V) release only after splicing is complete ( Figure 2—figure supplement 4 and ‘Materials and methods’). For each one of these general schemes, different time distributions were tested for elongation, splicing, and release ( Supplementary file 2 ). Since by construction, the intron-to-exon cross-correlation at 0 delay is necessarily null in scheme III and have a null slope in scheme I, these two schemes can be ruled out (See Figure 2—figure supplement 5A,D for fits). The three other schemes were better at fitting the correlation curves but the best model is one from scheme II, that is where splicing is independent of elongation and transcript release (See Figure 2—figure supplement 5 and discussion on Model comparison in ‘Materials and methods’). In this 3-parameter model ( Table 1 ), splicing occurs a fixed amount of time after the 3′ss has been transcribed, and transcript release involves a stochastic delay after the poly(A) site is reached. No pause at the 3′ ss was needed to fit the data. This observation does not rule out pausing at these sites but suggests that such a pause would be much shorter than the other timescales observed. Notably, our data are fit better with a model having a fixed time for intron removal rather than a stochastic (exponential) one, arguing for several sequential kinetic steps ( Aitken et al., 2011 ; Schmidt et al., 2011 ). In total, the β-globin-terminal intron splicing time was 267 ± 9 s after the polymerase passes the 3′ ss. This measurement of splicing time is consistent with previous estimates either in vivo on cell populations ( Singh and Padgett, 2009 ) or in vitro at the single-molecule level ( Hoskins et al., 2011 ), suggesting that PP7 stem loops do not perturb splicing kinetics of this intron, contrary to MS2 stem loops ( Aitken et al., 2011 ; Schmidt et al., 2011 ). As an independent validation of our modeling results, we counted the number of red and green RNAs at transcription sites using a normalized ratiometric approach ( Zenklusen et al., 2008 ) (See ‘Materials and methods’ and Figure 2—figure supplement 6 ). The average red-to-green ratio of 1.41 is indistinguishable from the expected 1.4 value predicted by our modeling analysis of the correlation functions.
Kinetics of transcription and splicing under different experimental conditions
The table shows result of fits with model II.4 (‘Materials and methods’ and Supplementary file 2 ). Pre-release fraction is deduced from the 3 other parameters. Errors are propagated SEM from correlation functions. * p-value<0.05, ** p-value<0.005 (two-sided z-test vs control).
Importantly, both SSA and CPT drug treatments only affected a single parameter ( Table 1 ), arguing that splicing is kinetically independent of elongation/termination. Based on our measurements, splicing occurring at the TS is rarely completed during elongation but rather during a pause at the 3′ end of the gene. Because transcript release is stochastic, the 3′ end dwell time can be shorter or longer than the necessary time to remove the intron, resulting in splicing that can occur either before or after release.
Splicing happens 10-fold faster on freely diffusing transcripts than on chromatin
Since the majority of β-globin pre-mRNA is released from the TS before splicing of the terminal intron, we addressed the question of where and when this splicing takes place. We observed a mobile population of unspliced pre-mRNA (co-localized intron/exon) diffusing in the immediate vicinity of the TS ( Figure 4A–B , Video 5 ). In contrast, spliced mRNA (exon only) could be observed diffusing throughout the nucleus. A small population of red-only particles was also recorded, which could be due either to the false-discovery rate of the segmentation algorithm or the presence of free lariats (See ‘Materials and methods’). The radial distribution of mRNA and pre-mRNA indicated an enrichment for unspliced transcripts within 2.4 ± 0.1 µm of the TS ( Figure 4C , Figure 4—figure supplement 1 ), meaning that splicing occurs faster than diffusion throughout the nucleus. This enrichment disappears upon treatment with splicing inhibitor SSA, in which case most of the transcripts are unspliced and dispersed throughout the nucleus ( Figure 4C , Video 6 ). From the measured diffusion coefficient (D = 0.12 µm 2 /s, Figure 4—figure supplement 2 ; See ‘Materials and methods’), we calculated that post-release splicing takes place on average 13 ± 1 s after departure from chromatin. This time is much shorter than the expected 137 s it would take if pre- and post-release splicing kinetics were identical (calculated from Table 1 ). From this observation, it is tempting to speculate that transcripts are released only after they have passed a particular rate-limiting step in spliceosome assembly, explaining why the catalytic step occurs very soon after release. However, this interpretation is inconsistent with the fact that 3′end retention time is not affected by SSA treatment which impairs binding of U2 and hence affects recruitment of all the snRNPs except U1 ( Corrionero et al., 2011 ). Note also that we cannot formally exclude the possibility that co-localized intron/exon particles are actually excised introns still in complex with transcripts. In summary, although introns can be retained for over 4 min on chromatin, once the transcript is released, splicing is 10-fold faster for freely diffusing transcripts. In most cases, the nascent intron is retained until transcription reaches the 3′ end of the gene and then removed either before or shortly after release of the transcript ( Figure 5 ).

Visualization of splicing occurring after release from chromatin.
( A ) Individual frames from live-cell confocal imaging showing intron (red dots), exon (green dots), and the merged image. White arrow: TS. Bar: 4 µm. ( B ) Fluorescence intensity profile along the line in the inset shows co-localized intron/exon (unspliced pre-mRNA) and exon only (spliced mRNA). ( C ) Radial distributions of mRNA (green) and pre-mRNA (orange), as well as pre-mRNA under SSA treatment (black) are shown as a function of distance from the TS. Density distributions are normalized by the distribution of random (uniform) positions within the nucleus (see ‘Materials and methods’). Error: SEM (bootstrap over 9 cells).

Single-RNA imaging reveals a transient population of unspliced transcript diffusing away from the transcription site.
Using high-power confocal laser scanning microscopy, we were able to observe single transcripts with a better temporal resolution than with widefield imaging ( Video 3 ). The video shows a single cell with an active transcription site (TS, bright spot visible in both signals) and diffusing RNA particles (left: intron, center: exon, right: merge). Although most of the RNAs diffusing in the nucleus are spliced (visible only in the exon signal), few unspliced RNAs (visible in both colors) are detectable in the vicinity of the TS as they diffuse away. Spatial distribution and diffusion analyses revealed that this population is very transient ( Figure 4C and Figure 4—figure supplements 1 and 2 ). Large shapes in the nucleus are nucleoli ( Ferguson and Larson, 2013 ).

Single-RNA imaging with splicing inhibitor SSA.
Imaging conditions are identical as in Video 5 , but cells are treated with splicing inhibitor spliceostatin A (SSA). RNAs diffusing in the nucleus are now visible in both color, indicating that they are unspliced.

Schematic of β-globin transcription cycle kinetics.
Transcript synthesis and processing can occur through different pathways, the choice of which is governed by a kinetic competition between transcription and splicing. After transcription of the 3′ splice site, intron removal takes about 260 s and elongation until the end of the gene, about 55 s. Hence, splicing does not occur during elongation. The transcript is retained at the 3′end of the gene for a stochastic amount of time that can be shorter or longer than the remaining time to excise the intron. This results in two possible outcomes: either an unspliced pre-mRNA is released and then spliced very rapidly or splicing occurs while the transcript is retained on chromatin before being released.
Recurrent cancer-associated mutation in splicing factor U2AF1 delays splicing and makes it entirely post-release
Because the balance of kinetic competition determines where and when introns are excised from the pre-mRNA, we then sought to determine whether trans -acting factors regulated splicing by perturbing this balance. Deep sequencing studies in myelodysplastic syndrome, chronic lymphocytic leukemia, acute myeloid leukemia (AML), breast cancer, lung adenocarcinoma, and hairy cell leukemia have all revealed the existence of mutated factors involved in 3′ ss recognition ( Yoshida et al., 2011 ; Graubert et al., 2012 ; Brooks et al., 2014 ; Waterfall et al., 2014 ; TCGA, 2012 ). One recurrent change is a heterozygous point mutation in U2 auxiliary factor 1 (U2AF1), which is an essential factor for recognition of the AG dinucleotide consensus motif ( Figure 1A ). The serine-to-phenylalanine (S34F) missense mutation in the zinc finger domain results in disparate changes in alternative splicing patterns including exon skipping, exon inclusion, and alternative 3′ ss selection ( Brooks et al., 2014 ). Although somatic genetics clearly indicate the importance of this mutation ( Waterfall et al., 2014 ), there is no functional or mechanistic understanding of how U2AF1-S34F works at the molecular level.
We performed time-lapse imaging on cells expressing moderate levels of either wild-type U2AF1 or U2AF1-S34F, both fused to a cerulean fluorescent protein. We note that this experimental condition recapitulates the in situ case, because the mutant U2AF1 is present against the background of at least one copy of the wild-type allele. Correlation functions revealed that U2AF1-S34F completely abolishes pre-release splicing ( Figure 6A , horizontal slope) and prolongs transcript 3′ end dwell time ( Table 1 ). Post-release imaging of transcripts showed a local enrichment for unspliced pre-mRNA near the TS, but with a greater spatial extent in the case of U2AF1-S34F ( Figure 6B–C , Video 7 ). Thus, all transcripts are post-transcriptionally spliced, albeit at a slower rate (27 ± 3 s; Figure 6D , Figure 4—figure supplement 1 ). Splicing efficiency, poly(A) length, and 3′ UTR length were unchanged ( Figure 1—figure supplement 1C–D) . In summary, these data suggest that the mutant delays splicing to post-release, slows splicing from freely diffusing transcripts, but has no detectable effect on splicing efficiency.

The U2AF1-S34F mutant acts as a dominant negative by delaying splicing to post-release.
( A ) Expression of U2AF1-cerulean does not alter pre-release splicing compared to the un-transfected control. Expression of U2AF1-S34F-cerulean abolishes splicing at the TS (horizontal slope of the intron-to-exon cross-correlation, blue curve). ( B ) Pre-mRNA (red, marked by squares) are enriched around the TS (arrows) indicating that splicing still occurs faster than diffusion. The enrichment is broader in the presence of U2AF1-S34F despite the similar spatial distributions of both proteins. ( C ) Gaussian fits onto pre-mRNA radial distance distributions from the TS. ( D ) The U2AF1-S34F mutant defers splicing to occur entirely away from the TS (fractions obtained from model fits in Table 1 ) and increases post-release splicing time. ** p < 0.005 (two-sided z-test vs untransfected control). ( E ) Two-color single-molecule FISH on endogenous FXR1 transcripts. Unspliced pre-mRNA (co-localization of intronic and exonic probe) appears in the vicinity of TSs (the 4 bright dual-color spots). ( F ) The fraction of pre-mRNA transcripts in the nucleus in the presence of wt or mutant U2AF1. ( G ) Spatial distribution of pre-mRNAs near TSs in the presence of wt or mutant U2AF1 (N > 400 cells). Radial distributions show density of pre-mRNA normalized by density of mRNA. Bars: 4 µm. Error: SEM over cells (bootstrap).

Spatial distribution of pre-mRNA with wild type or mutant U2AF1.
Left and right images show a cell that was transfected with the wild type (wt) or the mutant (S34F) version of splicing factor U2AF1. Both the image show the intron channel. The enrichment of unspliced pre-mRNA (red spots) diffusing in the vicinity of the transcription site is broader in the case of the mutant, showing that splicing rate is slower. Imaging conditions are identical as in Video 5 .
We then confirmed this kinetic effect on the endogenous FXR1 mRNA, which was shown to be alternatively spliced in the presence of U2AF1-S34F in both AML and lung adenocarcinoma ( Brooks et al., 2014 ). Using single-molecule FISH ( Femino et al., 1998 ) ( Figure 6E ), we examined the spatial distribution of intron and exon in fixed cells transfected with U2AF1 or U2AF1-S34F. As was the case for the β-globin reporter, we observed a population of unspliced FXR1 pre-mRNA in the vicinity of an active TS, indicating that at least some fraction of FXR1 transcripts are released before splicing ( Figure 6F ). We then performed the same spatial analysis as above, except on fixed cells instead of live cells. Expression of the U2AF1-S34F mutant resulted both in an increase in the level of unspliced pre-mRNA in the nucleus (16.0 ± 0.4% compared to 6.0 ± 0.3%, Figure 6F ) and the radial distance from the TS (1.8 ± 0.5 μm compared to 0.5 ± 0.2 μm, Figure 6G ). Taken together, both static and dynamic measurements on a reporter β-globin transcript and the endogenous FXR1 transcript suggest that the S34F mutation in U2AF1 acts in a dominant negative fashion to postpone splicing until after release and cause slower splicing from diffusing transcripts.
The picture that emerges from this study is one in which the β-globin-terminal intron can be spliced during multiple steps of the transcription cycle ( Figure 5 ). A minority of transcripts are spliced while retained at the 3′ end of the gene, but the dominant pathway is the one in which splicing is contemporaneous with release or occurs shortly thereafter. These results are consistent with several studies which suggested that intron removal is enhanced upon cleavage ( Baurén et al., 1998 ; Bird et al., 2005 ). In fact, we find that splicing is 10-fold faster on freely diffusing transcripts than on chromatin-bound transcripts. Our data are consistent with a model where commitment to splicing of the terminal β-globin intron occurs co-transcriptionally but suggest there may be a high energy barrier to completion of intron removal while transcripts are still tethered to chromatin, possibly due to steric constraints. Thus, post-release splicing may be the energetically favored process, but there is a long kinetic window in which the less-favorable pre-release intron removal can occur.
The time required to remove an intron therefore becomes a central parameter in our understanding of RNA processing, with implications for both constitutive and alternative splicing ( Bentley, 2014 ). This fundamental kinetic quantity has been elusive. Population-based measurements suggest a splicing time for several endogenous, un-modified introns of 5–10 min ( Singh and Padgett, 2009 ). Bulk in vitro measurements on β-globin indicate a 40- to 50-min timescale. A single-molecule in vitro study on yeast transcripts measured ∼10-min splicing time. On the opposite end of the spectrum, previous live-cell imaging approaches relying on both direct and indirect measurements indicate splicing times in the order of ∼30 s ( Huranová et al., 2010 ; Martin et al., 2013 ). One of the primary experimental advances in our study is the ability to observe the transcription and splicing process with a time resolution that spans three orders of magnitude. Since the time to splice is a distributed quantity, and splicing times vary from transcript to transcript, the variation in previously reported splicing rates may be strongly influenced by the temporal dynamic range of the method. Biologically, this variability in splicing rate may provide regulatory potential, as we discuss below.
One potential criticism of the live-cell imaging approach is that the stem-loops and the coat protein may perturb kinetics. Several arguments stand against this view. First, our splicing kinetics are consistent with both population measurements ( Singh and Padgett, 2009 ) and in vitro single-molecule measurements ( Hoskins et al., 2011 ). Second, the splicing efficiency of the reporter is high ( Figure 1—figure supplement 1C ), suggesting there are no dead-end intermediates. Third, endogenous metazoan RNAs are decorated with RNA-binding proteins from their inception ( Castello et al., 2012 ), suggesting that the spliceosome is well-equipped to handle bulky messages. Finally, since we can never exclude the possibility that a synthetic reporter may be missing features found in an endogenous gene, we have also recapitulated the results on the un-modified endogenous FXR1 message.
Importantly, our single-molecule study reveals the existence of multiple pathways, indicating the absence of a strict checkpoint for intron removal ( Bird et al., 2005 ; Alexander et al., 2010 ; Brody et al., 2011 ; Carrillo Oesterreich et al., 2010 ; Bhatt et al., 2012 ; Pandya-Jones et al., 2013 ). The presence of a delay at the 3′ end may be interpreted as a checkpoint mechanism to ensure that splicing takes place before transcript release, but there are several reasons to reject this interpretation of our data. First, none of the models which assume dependence between splicing and elongation/release fit better than one that assumes independence. Second, both splicing and elongation inhibition experiments support the view of the two processes being kinetically independent. Abolishing splicing with SSA does not lead to an increase in release times. Conversely, reducing elongation speed with CPT does not slow down splicing. Instead, it leads to an increase in pre-release splicing ( Figure 2E ), as expected if the two processes are independent. Thus, splicing and release can happen in either order, with the order of events determined by kinetic competition. It is this competition which results in stochastic outcomes for the RNA.
What are the physiological implications of this stochastic outcome? Does the cell utilize changes in kinetic balance to alter gene expression? Moreover, is the timing of intron removal of secondary importance to the timing of splicing commitment? While, single-cell variation in alternative splicing has been observed, the mechanism behind this variability has remained elusive ( Waks et al., 2011 ; Lee et al., 2014 ). We have found that a single point mutation in U2AF1 that is recurrent in multiple human cancers alters this kinetic balance to favor post-transcriptional splicing of both the β-globin reporter and also endogenous FXR1 mRNAs. Interestingly, single-molecule measurements on fixed cells suggest that post-release splicing may be the preferred pathway for alternatively spliced transcripts ( Vargas et al., 2011 ). Our studies on the mutant U2AF1 provide a mechanistic basis for this observation and suggest a role for mutations in the core splicing machinery for the increased levels of ‘noisy splicing’ which are observed in cancer ( Pickrell et al., 2010 ; Chen et al., 2011 ). Furthermore, our time-resolved results indicate that post-release splicing is more efficient than pre-release splicing, which may explain how the timing of intron removal might lead to different outcomes for the message. Such a model relies on a degree of plasticity in spliceosome assembly and function, which has been suggested by in vitro single-molecule measurements ( Hoskins et al., 2011 ; Shcherbakova et al., 2013 ). We speculate that the kinetic delay induced by U2AF1-S34F allows either for alternate pairing between 5′ and 3′ss during transcription or for post-release reconfiguration of pre-mRNA which is not yet committed to intron excision in the spliceosome. Other pathological RNA processing defects may also originate from a similar kinetic imbalance. In summary, the single-molecule approach developed here provides a blueprint for dissecting the many competing processes which take place at the earliest stages of gene expression.
Cell line and DNA constructs
The reporter gene vector ( Figure 1—figure supplement 1A ) was constructed with the human β-globin DNA sequence placed under the control of a Tet-responsive promoter, as described previously ( Janicki et al., 2004 ; Darzacq et al., 2007 ). Briefly, the 3′ end of the human β-globin sequence was truncated 72 bp upstream of the endogenous stop codon. It was replaced by a cassette coding for the cyan fluorescent protein fused to the peroxisome-targeting sequence serine-lysine-leucine (CFP-SKL), ending with a translation stop codon. A cassette containing 24 repeats of the MS2 stem loop sequence was inserted in the 3′ untranslated region (3′UTR), followed by a bovine growth hormone polyadenylation sequence (BGH-PolyA). In our modified construct, we inserted a cassette containing 24 repeats of the PP7 stem loop sequence into the second and last intron of the gene, approximately halfway between the 5′ splice site (5′ss) and the branch point. In order to be the least perturbing, the cassette was inserted 463 bp downstream of the 5′ss and 371 bp upstream of the branch point, and most of the endogenous DNA was conserved (8 bp in the intron were deleted). In addition, we replaced the MS2 cassette with one that is less prone to recombination. The repeating units of the PP7 and MS2 cassettes are composed of two-stem loop blocks, which are then multimerized 12× to get 24× total stem loops:
MS2: GATCCTACGGTACTTATTGCCAAGAAA GCACGAGCATCAGCCGTGC CTCCAGGTCGAATC
TTCAAA CGACGACGATCACGCGTCG CTCCAGTATTCCAGGGTTCATCAG
PP7: CTAAGGTACCTAATTGCCTAGAAA GGAGCAGACGATATGGCGTCGCTCC CTGCAGGTCGA
CTCTAGAAA CCAGCAGAGCATATGGGCTCGCTGG CTGCAGTATTCCCGGGTTCATTAGATC.The loops themselves are underlined.
The construct was stably integrated into the genomic DNA of U2-OS cells (human osteosarcoma cell line) through transfection of the plasmid followed by a screen for genomic integration. The cell line was made by co-transfection of the reporter plasmid and a puromycin resistance plasmid, followed by selection with puromycin. The cell line also constitutively expresses the PP7 bacteriophage-coat protein fused to the mCherry fluorescent protein (PP7-mCherry) and the MS2 bacteriophage-coat proteins fused to the green fluorescent protein (MS2-GFP). Both are under the control of a ubiquitin promoter and were stably introduced by lentivirus infection as described previously ( Larson et al., 2013 ). The clonal cell line used in this study was finally generated by single-cell cloning. The reporter is efficiently expressed and translated as evidenced from imaging of the protein product accumulating in peroxisomes ( Figure 1—figure supplement 1B ).
Cells were grown in DMEM medium (Life technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, St Louis, MO). Cell were induced with 10 or 20 μM doxycycline (Sigma Aldrich, St Louis, MO), at least 24 hr prior to imaging. Imaging was performed in Leibovitz L-15 phenol-free medium (Life technologies, Grand Island, NY) containing the same concentration of doxycycline and FBS. Pharmacological treatments were performed at 48 nM spliceostatin A (Dr Minoru Yoshida, Chemical Genetics Lab, RIKEN Institute, Japan) from 0.5 to 20 hr prior to imaging or 3.75 μM Camptothecin (Sigma, CAS: 7689-03-4) from 1 to 19hr.
U2AF1 (a.k.a. U2AF35) constructs were made from a U2AF35-CFP plasmid provided by Angus Lamond (University of Dundee, UK). The CFP cassette was replaced by a Cerulean one (similar absorption/emission spectra but more photostable). The U2AF1 cassette was then swapped with the mutant version (U2AF1-S34F) provided by Peter Aplan (NCI, NIH).
Splicing efficiency and poly(A) tail length/site
Cells were transfected with mock or with plasmids expressing either the WT or S34F mutant of U2AF1. Expression of β-globin reporter was induced for 15 hr. Cells were scraped and total RNA isolated using the Qiagen RNA isolation kit. 1 μg of total RNA was used to make first strand using ProtoScript II M-MuLV reverse transcriptase (NEB, Ipsich, MA) and random hexamers (IDT, Coralville, IA) according to the manufacturer's instructions in a final volume of 20 μl. 2 μl of the reverse transcription product was used in a qPCR reaction using IQ Syber Green mix (Bio-Rad, Hercules, CA) in a CFX96 qPCR machine (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. We use varying amounts of G-block DNA (IDT, Coralville, IA) carrying primer pairs generating amplicons of the same size as those being tested. This G-block served as control and was used to generate the standard curves. Primer pairs spanning the junction of Exon2-Intron2, and Intron2-Exon3, and a primer pair amplifying a region of Exon3 served to measure unspliced and total RNA of β-globin reporter respectively.
The unspliced fraction of the reporter RNA was calculated as 10^[ (Cq intron -K intron )/S intron - (Cq total -K total )/S total ] where Cq is the cycle number, and K and S are the constant and slope respectively of the standard curve for the primer pair ( Figure 1—figure supplement 1C ). The splicing fractions calculated using Exon2-Intron2 primers or Intron2-Exon3 primers were similar and were therefore combined together. Error bars are standard errors over four measurements.
U2-OS cells with the integrated β-globin reporter were transfected with mock or with plasmids expressing either the WT or S34F mutant of U2AF1. Expression of β-globin reporter was induced for 15 hr. Cells were scraped and total RNA isolated using the Qiagen RNA isolation kit. 1 μg of total RNA was treated with RNAse H (NEB, Ipswich, MA) in the presence oligo dT according to the manufacturer's instructions. The digested RNA was then phenol:chloroform extracted, ethanol precipitated and suspended in 10 μl of water. A DNA adaptor (5-GGTCACCTTGATCTGAAGC, with a 5-phosphate and 3 amino modification to prevent further ligation) was ligated to 1 μg of either total or RNAse H-treated RNA using T4 RNA ligase 1 (NEB, Ipswich, MA) in a final volume of 20 μl. 10 μl of the adaptor-ligated RNA was then used to synthesize the first strand using ProtoScript II RT (NEB, Ipswich, MA) and the reverse primer 5′-GCTTCAGATCAAGGTGACCTTTTT according to the manufacturer's instructions. The reaction was denatured for 20 min at 80°C and 2.5 μl of the first strand was used as template in a standard PCR reaction using the forward primer 5′-CCAGGGTTCATCAGATCCTATTCTATAGTGTCAC and the reverse primer 5-GCTTCAGATCAAGGTGACCTTTTT. Reaction products were then separated on a 3% agarose gel ( Figure 1—figure supplement 1D ).
Genomic integration sites and copy number analysis
Whole genome sequencing (paired end sequencing) was used to identify the integration sites of the reporter in the genome. The genomic library was prepared using TruSeq DNA sample preparation protocol. Samples were sequenced on HiSeq2000 using Illumina TruSeq v3 chemistry. The yield was 389 million reads after filtering. The reads were trimmed to remove low quality sequences (Trimmomatic software) and were aligned to both the human genome (hg19) and the sequence of the β-globin reporter gene plasmid (Bowtie2 and Illumina CASAVA Eland softwares). Paired reads that partially align to both genomic and plasmid sequences were extracted and used to identify possible insertion sites. Figure 1—figure supplement 2A shows an example of an insertion site: 4 read pairs align with both with genomic sequence (up to chr8:145,074,337) and plasmid sequence (all from the same position in the plasmid backbone). This defines the 5′ junction of the insertion. Similarly, three read pairs were found partially aligning a few bp downstream (chr8:145,074,349) and with the plasimd up to the beginning of the MS2 cassette, defining the 3′ junction. Multiple copies of the plasmid may have been integrated.
Three insertion sites were identified ( Figure 1—figure supplement 2B ), including two where only one junction was found. All three were confirmed by PCR and the copy number of the reporter in the cell line was obtained as follows. Genomic DNA was isolated from cells (1 × 10 cm plate) by standard protocols, and amplicons were PCR amplified using primer pairs overlapping the putative junctions. The PCR was performed for 24 cycles and the products separated on a 2% agarose gel ( Figure 1—figure supplement 2C ). All four junctions show a band at the expected size. To estimate the copy number of the inserted plasmid, we also use two primer pairs that amplify a region that is internal to the plasmid construct. Each junction should be present in one copy per cell so that, in comparison, the amount of the internal amplicon should reflect the total copy number in the cell line. The gel was quantitated using the ImageJ software ( Figure 1—figure supplement 2D ). To correct for primer efficiency, each primer pair was also used in the same PCR run to amplify varying amount of G-block DNA (IDT, Coralville, IA). The product was quantitated as described above and the resulting calibration curves ( Figure 1—figure supplement 2E ) were used to correct the data. The internal-to-junction ratio of the corrected amounts of PCR products indicates between 4 and 7 total copies of the reporter ( Figure 1—figure supplement 2F ).
Widefield microscopy and image processing
Microscopy data acquisition from Figure 1 , Figure 2 and Figure 6A,E–G (and associated supplements) and Videos 1 to 3 was performed using a custom build wide-field microscope (described in greater details in Ferguson and Larson, 2013 ). It consists in an AxioObserver inverted microscope (Zeiss, Thornwood, NY) with a high aperture objective (Zeiss 63× C-Apochromat) and two Evolve 512 EMCCD cameras (Photometrics, Tucson, AZ). Excitation sources are 488 nm and 594 nm lasers (Excelsior, Spectra Physics, Santa Clara, CA). Typical laser intensities are 250 μW and 50 μW respectively. For imaging Cerulean, we used a 445 LED (Zeiss Colibri) or an X-Cite lamp (Lumen Dynamics). Cells were imaged in 35-mm MatTek dishes (MatTek, Ashland, MA) placed in a Tokai Hit stage incubator (INUB-LPS, Shizuoka-ken, Japan). Average temperature inside the dish was measured at 37°C using a thermocouple. Images were taken every 10 s as z-stacks (7 or 9 images, Δz = 0.5 μm) and in both color simultaneously with two cameras, using an exposure time of 100 ms and for a duration between 45 and 512 frames. Raw images were collected using MicroManager software ( Edelstein et al., 2001 ). Maximum intensity projections were computed (e.g. Video 1 ) and used for tracking. Video 3 was obtained using a shorter exposure time (50 ms) to observe the fluctuations due to diffusing RNAs.
Bicolor fluorescence time traces at the transcription site were generated using a custom software written in IDL ( Source code 1 ) that was previously described, with minor modifications to handle bicolor data sets. In each image, diffraction-limited spots are detected using band-pass filtering and refined using an iterative Gaussian mask localization procedure ( Crocker and Grier, 1996 ; Thompson et al., 2002 ; Larson et al., 2005 , 2011 ). Trajectories are then generated based on a nearest-neighbor method with a maximal jump distance threshold. If no spot is detected within the threshold distance, the previous location is used as the initial guess for the iterative Gaussian mask localization procedure. Integrated fluorescence intensity over the diffraction-limited spot is collected using a Gaussian mask fit after local background subtraction ( Thompson et al., 2002 ). An example of time-lapse video with tracking is shown in Videos 2 .
Time traces were corrected for photobleaching as follows. In an ideal experiment, the fluorescence intensity histogram of the whole nucleus should stay roughly unchanged throughout the acquisition. We computed smoothed versions (polynomial fit) of the mean and standard deviation (s.d.) of this histogram in each color over time. Time traces from TS tracking were then normalized by the s.d. (because of the background subtraction in the tracking procedure, only the s.d. of the nucleus histogram acts as a scale factor on a time trace).
Traces were inspected for (i) accurate tracking (portions of inacurate tracking were trimmed off; short traces (<100 frames) were discarded), (ii) in-focus TS (traces where TS reaches the first or last z-plan were discarded), and (iii) signal to noise ratio (highly noisy traces were discarded). Examples are shown on Figure 1C and Figure 2—figure supplement 1A .
Fluctuation analysis
For each time trace, autocorrelation and crosscorrelation functions were computed as
where <·> denotes time average, δa(t) means a(t) - áa(t)ñ, and a(t) and b(t) can be any combination of the red and green time traces r(t) and g(t).
Correlation functions were computed using a multi-tau algorithm ( Wohland et al., 2001 ), which iteratively down-samples the signals for increasing time delay. This yields a somewhat uniform spacing of time delay points on a logarithmic scale, reducing the sampling noise at long delays while keeping a high temporal resolution at short delays. When shifting the two signals, non-overlapping ends are not wrapped. See Figure 2—figure supplement 1A for examples of correlation functions from single traces.
To reach better statistical convergence, correlation functions from single time traces were averaged together ( Figure 2—figure supplement 1B ). Each point of the single-trace correlation functions was given a weight corresponding to the number of overlapping time points from the signals used in its computation. Bootstrapping was performed to obtain standard error of the mean correlation functions (SEM). Two normalizations were performed prior to bootstrapping: (i) Baseline subtraction: Slow processes (e.g. bursting, cell cycle) may produce a slow decay which adds up to the fast transcription/splicing kinetics. These are usually well separated; see how the decay is much slower after 200–300 s on Figure 2—figure supplement 1B–F . The baseline subtraction gets rid of the slow decay, approximating it as a constant offset at short delays. (ii) Normalization with null-delay crosscorrelation G rg (0): All 4 correlation functions were normalized using an estimate of G rg (0). This was done for 2 reasons: (a) doing this normalization prior to bootstrapping promotes a good convergence of the crosscorrelation at short delays 0 (the most informative part of the correlation functions; See Supplementary file 1 —§2). This constrains the fits to capture precisely the temporal features at short delays. (b) This normalization reduces by 1 the number of free parameters of all the models used to fit the data: On unnormalized correlation functions, changing the initiation rate of transcription simply scales up and down all 4 correlation functions together (See Supplementary file 1 —§1). Normalizing them gets rid of this degree of freedom. Because most of the time G rg (0) is inaccurate due to shot noise and/or small tracking error (e.g. Figure 2—figure supplement 1C,E,F ), we use the estimate (G rg (Δt) + G gr (Δt))/2 instead (Δt = 10 s is the sampling time).
Confocal microscopy and image processing
We performed imaging with a confocal laser scanning microscope to observe single RNAs diffusing in the nucleus ( Figure 4 , Figure 6B–C [associated supplements], Figure 2—figure supplement 6 and Videos 5 to 7 ). Imaging was performed on a Zeiss 780 confocal microscope with 488/594 nm excitation.
To detect and localize RNAs in the nucleus, we used the same spot-localization algorithm as for widefield microscopy ( Figure 4—figure supplement 1B ). False positive spots due to nucleoli or localized outside of the nucleus were eliminated using a standard masking procedure. Spots were detected independently in both channels (intron and exon) in 9 videos of 50 frames on average taken with a 3.26 s frame interval. Pairs of red and green spots colocalized by less than 250 nm ( Figure 4—figure supplement 1C ) were hence considered single bi-color particles. The TS was tracked and used as a reference for radial distributions of particles ( Figure 4C , Figure 4—figure supplement 1D ). These distributions were normalized by the distribution obtained for random locations in the nucleus (i.e., a value of 1 observed indicates a purely uniform distribution). RNAs in the nucleus being relatively sparse, colocalized particles are expected to be mostly true positive, that is unspliced RNAs. On the other hand, a proportion of the red-only and green-only particles may be false positive (e.g., bi-color RNA only detected in one color). mRNAs being much more abundant than pre-mRNA ( Figure 4A ), green-only spots should only have a small portion of false positives. It is however difficult to estimate what portion of the red-only spots truly represents free lariat and what is a wrong detection/categorization of pre-mRNA. The fact that the red-only radial distributions and the co-localized radial distribution are very similar suggests that red-only spots have a high proportion of false positives ( Figure 4—figure supplement 1D ) and that lariat lifetime is likely shorter than that of post-release pre-mRNA (i.e. < 13 s). Depletion at distances shorter than 1 μm is due to the difficulty for the spot-localization algorithm to locate two spots closer than this distance. The radial distribution of colocalized red–green spots was fit to a Gaussian distribution with three parameters: standard deviation σ , height h , and baseline y 0 ( Figure 4—figure supplement 1E ).
We determined the diffusion coefficient of RNAs in the nucleus using raster image correlation spectroscopy (RICS) ( Brown et al., 2008 ) ( Figure 4—figure supplement 2 ). Imaging was performed in photon counting mode. Time series of 10 frames and 512 × 512 pixels were taken with a 100 μs pixel dwell time, 61 ms line scanning time, and 52 nm pixel size. The 1/e2 beam waist was determined to be 246 nm in the green channel and 375 nm in the red channel by fitting a 2D Gaussian to a profile through the diffraction limited transcription site. Correlation functions were calculated and fit to a two component diffusion model using the Globals software package developed at the Laboratory for Fluorescence Dynamics at the University of California at Irvine ( http://www.lfd.uci.edu/globals/ ).
Based on the distribution of unspliced transcripts around the TS (i.e., normal distribution with σ = 2.421 μm ± 0.087) and a diffusion coefficient of D = 0.12 μm 2 /s, we deduce that the splicing time following release is on average σ 2 /4D = 12.71 s ± 0.92. We applied the same analysis to confocal videos obtained on cells transfected with wild-type or mutant version of U2AF1 ( Figure 4—figure supplement 1E ).
We also used confocal images to normalize the intensity of the transcription site by the intensity of single transcripts, allowing us to count the number of nascent transcripts in both channels ( Figure 2—figure supplement 6 ). The results were in agreement with the transcription and splicing kinetic parameters we found in our modeling analysis ( Table 1 ).
Fluorescence in situ hybridization (FISH)
To confirm our results obtained on a reporter gene, we performed FISH on the endogenous gene FXR1 (fragile X mental retardation). FXR1 was identified by Brooks et al. (2014) as being alternatively spliced in the presence of U2AF1-S34F mutation (i.e., higher retention of the second to last exon). According to our whole genome sequencing data (See above), FXR1 is at a tetraploid locus in our line of U2-OS cells.
We designed 48 probes in the second to last intron (right upstream the alternatively spliced exon) and 48 probes in the exons of FXR1 that are common between all the RefSeq variants, and excluding the two last exons ( Figure 6E ). Probes were generally 20 nucleotides. Intronic and exonic probe sets were synthesized and labeled with cyanine dyes Cy3 and Cy5, respectively, by Biosearch Technologies (Petaluma, CA). U2-OS cells from the same cell line as in the rest of our study were grown on coverslips and transfected with wild type (wt) or mutant (S34F) U2AF1 labelled with Cerulean fluorescent protein, 24 hr prior to fixation. Fixation and hybridization were performed according to the Stellaris RNA FISH protocol (Biosearch Technologies, Petaluma, CA). Coverslips were mounted on microscope slides using mounting media with DAPI (ProLong Gold antifade reagent, Life Technologies).
Imaging was performed on the same widefield microscope as described above. Light sources for imaging DAPI, Cy3, Cy5, and Cerulean were 365 nm, 530 nm, 625 nm, and 445 nm LEDs respectively, from a Zeiss Colibri (Zeiss, Thornwood, NY). The detector was a Hamamatsu ORCA-R2 C10600 camera (Hamamatsu Photonics, Japan). Fields of view were selected for cells with low levels of U2AF1-Cerulean and stacks of nine images with z-step of 0.5 μm were acquired in four colors. Maximum intensity projection images were used for analysis. Diffraction limited spots were identified in the Cy3 and Cy5 channels independently using the same software as above. Nuclear masks were generated from the DAPI channel using CellProfiler software (Broad Institute, http://www.cellprofiler.org ). Spots within 200 nm of each other were paired and considered a bicolor particle. Bicolor particles with a fluorescence more than a twofold of that of single RNAs were considered transcription sites (TS).
The measured number of mRNA (exon only particles) was similar with wild-type U2AF1 (29.9 mRNA/nucleus) and with the mutant (35.5 mRNA/nucleus). However, the fraction of pre-mRNAs over total nuclear RNAs was significantly different: 6.0% (±0.3) with the wild type and 16.0% (±0.4) with the mutant ( Figure 6F ). Radial distributions were obtained by computing the distance between single RNAs and all the TSs in a nucleus. Normalized density shown in Figure 6G is the density of pre-mRNA over the density of mRNA. The enrichment at short distances was fit with a Gaussian distribution as for the confocal data. The standard deviation parameter reveals that the local enrichment for pre-mRNA near TSs is broader with U2AF1-S34F (1.83 ± 0.49 μm) than with U2AF1-wt (0.48 ± 0.19 μm). Note that the non-null baseline in the radial distributions also reveals a second population of pre-mRNAs which are spliced slower than diffusion in the nucleus. The proportion of this second population as well as its spatial distribution is not affected by U2AF1-S34F mutation.
Mechanistic models for RNA synthesis and processing
In Supplementary file 1 —§2, we showed how different features in the geometry of the correlation functions (e.g., position of specific angles, decay times, change of slope, …) are reflecting different aspects of the transcription/splicing kinetics (e.g., elongation speed, intron and exon dwell times, stochastic co- and post transcriptional splicing, …).
Here, we want to build mathematical models that fully predict the correlation functions given (i) a certain underlying mechanism and (ii) a set of parameters. In essence, we want to ask what molecular mechanisms are consistent with all the above-mentioned kinetic features—and possibly more—that the correlation functions are encoding. The caveat is however that these models necessarily make assumptions on the underlying mechanisms. Hence, we take a general approach in generating a series of simple models (each assuming different mechanisms) and assess which one(s) account for the experimental data.
Generic approach
To generate a series of 21 minimalistic competing models, we adopt a strategy in two steps. The first step consists in formulating five different general mechanistic schemes, each assuming a different causality relationship between transcription and splicing ( Figure 2—figure supplement 4 ):
•scheme I assumes that splicing is entirely post-release,
•scheme II assumes no interdependence between splicing and transcription/release,
•schemes III, IV, and V assume three different checkpoint mechanisms between transcription and splicing (i.e., one process waits for the other).
These schemes are formulated in a generic way by describing the delays between key events in the transcription/splicing process as arbitrary distributions A ( t ), B ( t ), C ( t ), D ( t ), E ( t ), and F ( t ), as shown on Figure 2—figure supplement 4 . Using this generic description, we are able to derive the general expressions of the correlation functions for each scheme ( Supplementary file 1 —Appendix 2). The second step is to generate multiple simple models from each of these schemes by affecting A ( t ), B ( t ), C ( t ), D ( t ), E ( t ), and F ( t ) with particular distributions. For instance, in a model where everything is deterministic, all the distributions will be Diracs. If a certain step includes a stochastic pause, it will involve an exponential distribution. If a step results from the succession of several biochemical reactions, it will involve a gamma distribution. Supplementary file 2 summarizes all the different models we derived from each scheme.
For each of these models (described thereafter), changing the parameter value(s) produces different sets of correlation curves. We fit these correlation curves to the experimental data (see examples for the untreated condition on Figure 2—figure supplement 5A–F ) by fitting all four experimental correlation functions at once. We used a Levenberg-Marquardt non-linear least square fitting procedure, giving twice more weight to the crosscorrelations since they carry more information. Only time points up to typically 350∼500 s were used. Fit quality was compared between models using a Bayesian Information Criterion (BIC, See Supplementary file 1 —Appendix 3) on Figure 2—figure supplement 5 (the lower the BIC, the better the fit). The BIC favors simpler models: if two models with different number of parameters fit equally well the data, the BIC penalizes the one with extra degrees of freedom so that only the simplest model is retained.
In addition, as indicated in Supplementary file 2 , each model may or may not be consistent with:
•the geometrical properties of G rg (0) described in the previous section ( Figure 2—figure supplement 2C–D ; example, scheme I necessarily yields a flat G rg (0 + ) by construction), or
•the fact that transcripts can be released unspliced from the transcription site ( Figure 4A–B ).
Details of schemes and models
Schematics of each scheme are shown on Figure 2—figure supplement 4 and the models generated from them are listed in Supplementary file 2 . The general forms of the correlation functions are derived in Supplementary file 1 —Appendix 2.
Scheme I—splicing is post-release
Here, the red signal always remains as long as the RNA is being transcribed and drops coincidentally with the green signal, when the transcript is released. Model I.1 is the simplest possible: it assumes that everything is deterministic, that elongation proceeds with a constant speed, and that transcript release is instantaneous. It has a single parameter v, representing the speed of the polymerase. In models I.2, we add an elongation pause between the two cassettes. This can represent a pause anywhere between the cassettes, such as, in particular, right after the 3′ splice site (3′ss). This model has two parameters: the elongation speed v and the mean pause time ps. Model I.3 is constructed similarly by introducing an exponentially distributed pause at the 3′ end of the gene. Model I.4 includes pauses both after the 3′ss and at the 3′ end of the gene.
Scheme II—no crosstalk between transcription and splicing
Once the 3′ss is passed, splicing and transcription proceed independently and may complete in either order for different transcripts as a result of their own stochastic kinetics. The red signal drops either when splicing occurs (after F ( t )) or when the transcript is released (after E ( t )), whichever of the two happens first. Models II.1 and II.2 consider a deterministic elongation (equivalent of model I.1) with either a deterministic or a stochastic splicing time. Models II.3 to II.5 include a pause at the 3′ss, at the 3′ end of the gene, or at both (as for models I.2 to I.4), but also include here a deterministic splicing time. Of all the models described so far, the experimental data is best fit by model II.4 where splicing is deterministic and 3′ end dwell time exponential. Hence, we generated three other closely related models: II.6 where splicing is exponential and 3′ end dwell time deterministic, II.7 where both are exponential, and II.8 where 3′ end dwell time is exponential and splicing follows a gamma distribution.
Scheme III—checkpoint at 3′ splice site: Elongation pauses after the 3′ss until intron is spliced
This is an obligatory checkpoint because splicing has to complete before the polymerase keeps elongating, as opposed to models II.3 and II.5 where there is a pause after the 3′ss regardless of the splicing kinetics. By construction, the red signal always drops before the green signal starts rising. Hence, G rg (0) is necessarily null and flat on both sides of the y-axis (first case of Figure 2—figure supplement 2C ), making it obviously unable to fit our experimental data. For the sake of illustration, we derived two models, respectively with an exponential and a deterministic splicing/pausing time. Both fit very badly the experimental data ( Figure 2—figure supplement 5D ).
Scheme IV—checkpoint at exon end/termination site
Splicing can only take place after the polymerase has reached the termination site (or the 3′ end of the last exon). Once splicing happens, release of the transcript may require an extra time (distribution F ( t )). In models IV.1 to IV.3, when the polymerase reaches the termination site, splicing occurs either immediately (IV.1), or after an exponentially (IV.2) or gamma distributed (IV.3) delay. Then, the RNA is released after an exponentially distributed time (e.g., for additional 3′ end processing). These three cases correspond to a checkpoint mechanism that holds the splicing process respectively at its last biochemical step, or at 1 or several steps prior to its completion. In this scheme, RNAs cannot be released unspliced. So the only possibility for G rg (0) to show both a break of slope and a rising slope at 0 + would be that splicing and release occur simultaneously for a fraction of the transcripts. Hence, we built model IV.4 which is similar to model IV.2 except that, for a fraction of the transcripts, release is immediate after splicing. This corresponds to a mechanism where both splicing and RNA 3′-end processing take (independent) exponential times after the termination site is reached, but the RNA is only released when both processes have come to completion.
Scheme V—checkpoint for release
After the polymerase has passed the 3′ss, elongation and splicing happen independently of each other as in scheme II. However, here, the transcript can only be released after the intron is removed. In models V.1 to V.3, the splicing time is respectively deterministic, exponentially distributed, or follows a gamma distribution, corresponding to different number of biochemical reaction in the splicing process.
Model comparison
Evaluating all the models on each experimental data set ( Figure 2—figure supplement 5 ) using the Bayesian Information Criterion (BIC, Supplementary file 1 —Appendix 3), we find that, the most likely models are either II.4 or IV.4 in the control case, as well as in many of the other cases. Models IV.3 and V.3 perform well in a number of cases, but only V.3 is consistent with all the geometric features of G rg (0) ( Supplementary file 2 ), making it also a good candidate to explain the data. Complementary to the BIC, a visual inspection of the fits ( Figure 2—figure supplement 5A–F ) also illustrates how the different models perform. In particular, how badly models II.1 to II.3 fit the data indicates the need for a pause at 3′ end.
To conclude our modeling and data fitting analysis, three candidate models are able to reproduce the data accurately: II.4, IV.4, and V.3. All three models agree on the following:
•the delay between splicing and release is stochastic,
•only a fraction of the RNAs are spliced strictly before release,
•pre-release splicing occurs mostly (or exclusively) at the 3′ end of the gene,
•all three models give similar values for delays between events at the transcription site (so that any may be used to report these delays).
These models differ on the causality between events: whether splicing and transcription are independent or one waits for the other cannot be known purely from the correlation curves themselves. Model II.4, however, appears as the simplest of all three since it assumes independence instead of a checkpoint mechanism and has only three parameters. In addition, several strong lines of evidence point toward model II.4. Indeed, only this model is consistent with the following observations:
•inhibiting splicing does not affect 3′-end dwell time ( Table 1 ),
•inhibiting elongation does not affect splicing time ( Table 1 ),
•some unspliced RNAs can be released from the transcription site ( Figure 4 and Video 5 ).
These observations argue that splicing and transcription are independent. Hence, we conclude that model II.4 is the most likely mechanism and use it in the main article to report parameter values.
Error on model parameters and statistical tests
We used error propagation from the correlation functions to obtain SEM on the fitting parameters. To assess statistical significance in Table 1 , a standard two-sided z-test was systematically performed, for each model parameter, between the untreated condition and all the other conditions.
- Alexander RD
- Google Scholar
- Obtulowicz T
- Karkusiewicz I
- Mariconti L
- Tollervey D
- Wieslander L
- Chartrand P
- Pandya-Jones A
- Farabaugh S
- de la Mata M
- Knezevich A
- Pradet-Balade B
- Kornblihtt A
- Bieberstein N
- Böhnlein EM
- Neugebauer KM
- Sharifnia T
- Imielinski M
- Pedamallu CS
- Sivachenko A
- Rosenberg M
- Chmielecki J
- Lawrence MS
- Buenrostro JD
- Greenleaf WJ
- Carrillo Oesterreich F
- Preibisch S
- Eichelbaum K
- Beckmann BM
- Humphreys DT
- Steinmetz LM
- Krijgsveld J
- Patskovsky Y
- Tovar-Corona JM
- Churchman LS
- Weissman JS
- Waterfall JJ
- Corrionero A
- Valcárcel J
- de Turris V
- Blaustein MA
- Kornblihtt AR
- Natarajan P
- Drinnenberg IA
- Schiller BJ
- Yates JR III
- Edelstein A
- Glover-Cutter K
- Zawilski SM
- Graubert TA
- Okeyo-Owuor T
- McLellan MD
- Kalicki-Veizer J
- O'Laughlin M
- Tomasson MH
- Westervelt P
- DiPersio JF
- Zenklusen D
- Darnell JE Jnr
- Friedman LJ
- Gallagher SS
- Crawford DJ
- Anderson EG
- Wombacher R
- Tsukamoto T
- Salghetti SE
- Sachidanandam R
- Prasanth KV
- Motoyoshi H
- Horinouchi S
- Lawrence DS
- Windgassen T
- Irudayaraj J
- Kirchhausen T
- Carmo-Fonseca M
- dos Santos G
- Pickrell JK
- Pritchard JK
- Villemin JP
- Shcherbakova I
- Corrêa IR Jnr
- Thompson RE
- Levandoski M
- Kreitman RJ
- Shiraishi Y
- Sato-Otsubo A
- Chalkidis G
- Yamaguchi T
- Sakata-Yanagimoto M
- Haferlach C
- Koeffler HP
- Haferlach T
- Rosenfeld L
Author details
Contribution, contributed equally with, competing interests, present address.
For correspondence
National cancer institute (1ziabc011383-03), national institute of diabetes and digestive and kidney diseases.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors would like to thank Tom Johnson and Joseph Rodriguez for assistance with qRT-PCR and Nico Sturman and Arthur Edelstein for help with μManager. Spliceostatin A was a kind gift from M. Yoshida, RIKEN ASI. U2AF1 (wt and S34F) were provided by Angus Lamond and Peter Aplan. Aaron Hoskins and Joseph Rodriguez provided critical feedback on the manuscript. The cell line was sequenced by the sequencing facility of NCI, Frederick. The authors would like to acknowledge initial support from R01GM086217 to Robert Singer. The authors thank members of the Janelia Farm/HHMI Transcription Imaging Consortium for helpful discussions.
Version history
- Received: July 11, 2014
- Accepted: October 1, 2014
- Accepted Manuscript published: October 1, 2014 (version 1)
- Version of Record published: October 28, 2014 (version 2)
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication .
- 8,636 views
- 1,382 downloads
- 142 citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
Downloads (link to download the article as pdf).
- Article PDF
- Figures PDF
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools), categories and tags.
- transcription
- RNA processing
- single-molecule imaging
- fluctuation analysis
Research organism
Further reading.
- Cell Biology
Heat stress impairs centromere structure and segregation of meiotic chromosomes in Arabidopsis
Heat stress is a major threat to global crop production, and understanding its impact on plant fertility is crucial for developing climate-resilient crops. Despite the known negative effects of heat stress on plant reproduction, the underlying molecular mechanisms remain poorly understood. Here, we investigated the impact of elevated temperature on centromere structure and chromosome segregation during meiosis in Arabidopsis thaliana . Consistent with previous studies, heat stress leads to a decline in fertility and micronuclei formation in pollen mother cells. Our results reveal that elevated temperature causes a decrease in the amount of centromeric histone and the kinetochore protein BMF1 at meiotic centromeres with increasing temperature. Furthermore, we show that heat stress increases the duration of meiotic divisions and prolongs the activity of the spindle assembly checkpoint during meiosis I, indicating an impaired efficiency of the kinetochore attachments to spindle microtubules. Our analysis of mutants with reduced levels of centromeric histone suggests that weakened centromeres sensitize plants to elevated temperature, resulting in meiotic defects and reduced fertility even at moderate temperatures. These results indicate that the structure and functionality of meiotic centromeres in Arabidopsis are highly sensitive to heat stress, and suggest that centromeres and kinetochores may represent a critical bottleneck in plant adaptation to increasing temperatures.
Post-transcriptional splicing can occur in a slow-moving zone around the gene
Splicing is the stepwise molecular process by which introns are removed from pre-mRNA and exons are joined together to form mature mRNA sequences. The ordering and spatial distribution of these steps remain controversial, with opposing models suggesting splicing occurs either during or after transcription. We used single-molecule RNA FISH, expansion microscopy, and live-cell imaging to reveal the spatiotemporal distribution of nascent transcripts in mammalian cells. At super-resolution levels, we found that pre-mRNA formed clouds around the transcription site. These clouds indicate the existence of a transcription-site-proximal zone through which RNA move more slowly than in the nucleoplasm. Full-length pre-mRNA undergo continuous splicing as they move through this zone following transcription, suggesting a model in which splicing can occur post-transcriptionally but still within the proximity of the transcription site, thus seeming co-transcriptional by most assays. These results may unify conflicting reports of co-transcriptional versus post-transcriptional splicing.
- Genetics and Genomics
Single-cell ‘omic profiles of human aortic endothelial cells in vitro and human atherosclerotic lesions ex vivo reveal heterogeneity of endothelial subtype and response to activating perturbations
Heterogeneity in endothelial cell (EC) sub-phenotypes is becoming increasingly appreciated in atherosclerosis progression. Still, studies quantifying EC heterogeneity across whole transcriptomes and epigenomes in both in vitro and in vivo models are lacking. Multiomic profiling concurrently measuring transcriptomes and accessible chromatin in the same single cells was performed on six distinct primary cultures of human aortic ECs (HAECs) exposed to activating environments characteristic of the atherosclerotic microenvironment in vitro. Meta-analysis of single-cell transcriptomes across 17 human ex vivo arterial specimens was performed and two computational approaches quantitatively evaluated the similarity in molecular profiles between heterogeneous in vitro and ex vivo cell profiles. HAEC cultures were reproducibly populated by four major clusters with distinct pathway enrichment profiles and modest heterogeneous responses: EC1-angiogenic, EC2-proliferative, EC3-activated/mesenchymal-like, and EC4-mesenchymal. Quantitative comparisons between in vitro and ex vivo transcriptomes confirmed EC1 and EC2 as most canonically EC-like, and EC4 as most mesenchymal with minimal effects elicited by siERG and IL1B. Lastly, accessible chromatin regions unique to EC2 and EC4 were most enriched for coronary artery disease (CAD)-associated single-nucleotide polymorphisms from Genome Wide Association Studies (GWAS), suggesting that these cell phenotypes harbor CAD-modulating mechanisms. Primary EC cultures contain markedly heterogeneous cell subtypes defined by their molecular profiles. Surprisingly, the perturbations used here only modestly shifted cells between subpopulations, suggesting relatively stable molecular phenotypes in culture. Identifying consistently heterogeneous EC subpopulations between in vitro and ex vivo models should pave the way for improving in vitro systems while enabling the mechanisms governing heterogeneous cell state decisions.
Be the first to read new articles from eLife


Krasnodar, Russia

Tours, Attractions and Things To Do in Krasnodar
Here we are in the South of Russia. Welcome to Krasnodar, a major economic and cultural center of North Caucasus, also called “the capital of Kuban”. Krasnodar, perhaps, is one of the most interesting resort towns of the Russian Federation. It is a relatively young city founded by the Cossacks in в 1793 on the lands granted by Ekaterina II (hence its former name was Ekaterinodar). Later, when the Soviet system rose to power, the city was re-named to Krasnodar in 1920, and it preserved this name up to date.
Krasnodar is a center of the Russia’s southern touristic zone, located of the right bank of the Kuban River, 120-150 km from two warm seas – the Black and the Azov. It is an interesting fact that the city is located in the golden section of the Earth, almost in between the equator and the North Pole, right on the 45th parallel, also called “the Golden Line” or “the Life Line”. There exists an opinion that the living conditions in these latitudes are most favorable for human.
In spite of the city’s “youth” Krasnodar has many historical landmarks, while its architectural look is various and represented by different styles from Baroque and Classicism to late Modern. In Krasnodar, there is one of the largest Russian churches – Saint Catherine’s Cathedral, built as early as in 1914 and survived by a miracle under the Soviet power. It is also worth while visiting the Krasnodar main Orthodox Church – Alexander Nevsky Cathedral. Interesting and dramatic, was the fate of this, one of the Russia’s most beautiful churches, an example of Russian and Byzantine templar style. Alexander Nevsky Military Cathedral was erected in April 1853, but later on, in year 1932 it was blown up by the Communists. And it was not until May 2006 when the inauguration ceremony of Alexander Nevsky Cathedral built anew took place.
Nowadays the Krasnodar architectural variety combines the harmony of old and modern structures. Now, next to the churches there are also modern high-rise buildings (the Marriott Hotel”), shopping and entertainment centers (“Red Square”), modern concert halls (Palace of Arts “Premiere”), restaurants, and night clubs. The city also strikes with its numerous museums, theaters, art galleries. Particularly, if you find yourself in Krasnodar, we recommend you to visit the Krasnodar Regional Art Museum named after F. A. Kovalenko to enjoy a rare collection of Russian avant-garde and Dutch art of XVI century, and also attend concerts of SSAI “Kuban Cossack chorus” to listen to Kuban Cossack, Russian and Ukrainian folk-songs.
Two Krasnodar unusual landmarks enjoy wide popularity among tourists and local community: the Monument to a purse and the Monument to the doggies in love. Do you want to turn round? Then you are to the purse. It is enough to rub your purse over it, and you will have more money. But if you are unlucky in love, then the loving couple of dogs will help you, you should stroke their small paws and love will certainly come to you.

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Cardiovasc Endocrinol Metab
- v.9(4); 2020 Dec
Pathogenesis and remission of type 2 diabetes: what has the twin cycle hypothesis taught us?
Ahmad al-mrabeh.
Magnetic Resonance Centre, Translational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, UK
Type 2 diabetes has been regarded a complex multifactorial disease that lead to serious health complications including high cardiovascular risks. The twin cycle hypothesis postulated that both hepatic insulin resistance and dysfunction rather than death of beta (β) cell determine diabetes onset. Several studies were carried out to test this hypothesis, and all demonstrated that chronic excess calorie intake and ectopic fat accumulation within the liver and pancreas are fundamental to the development of this disease. However, these recent research advances cannot determine the exact cause of this disease. In this review, the major factors that contribute to the pathogenesis and remission of type 2 diabetes will be outlined. Importantly, the effect of disordered lipid metabolism, characterized by altered hepatic triglyceride export will be discussed. Additionally, the observed changes in pancreas morphology in type 2 diabetes will be highlighted and discussed in relation to β cell function.
Introduction
Type 2 diabetes (T2DM) has become a global concern. According to the WHO and International Diabetes Federation reports, 425 million of the world population have diabetes, and this is expected to double in the next few decades due to the large number of people at prediabetes stage [ 1 , 2 ]. These figures are alarming, and would place a major burden on national health systems across world. In the UK alone, there are 3.8 million people diagnosed with diabetes, and T2DM is currently costing 10% of the National Health System budget [ 3 ].
T2DM is a slow onset disease that develops over many years of insulin resistance and progressive decline of β-cell function [ 4 ]. Despite contribution of genetic and environmental factors [ 5 ], metabolic factors are critical in determining the onset of T2DM. Excess calorie intake over many years is the factor that triggers ectopic fat storage and subsequent derangement in lipid metabolism. The latter will lead to a number of cellular processes that limit hepatic responsiveness to insulin function and decreased β-cell function. In the early years of disease development, blood glucose levels remain normal due to the high compensatory ability of the β-cell to encounter insulin resistance. Hence, it is unlikely that high glucose is the initiating factor of β-cell damage. Eventually, the continued fat-driven impairment of β-cell function will lead to the development of T2DM when approximately 40–60% of β-cell functional mass is lost [ 6 – 8 ].
For many years, T2DM has been considered a chronic disease, which is inevitably progressive. However, a series of studies – Counterpoint, Counterbalance and the Diabetes Remission Clinical Trial (DiRECT) – have changed this view. It has been demonstrated that long-term remission of T2DM is achievable via effective diet-induced weight loss [ 9 – 12 ]. The underlying pathophysiologic mechanisms that determine diabetes development are now broadly understood. The twin cycle hypothesis was published in 2008 to predict the aetiology and pathophysiology of T2DM development and reversal [ 13 ]. This was tested in several studies [ 9 , 10 ], and all emphasized the role of excess fat within the liver and pancreas on the pathogenesis of this disease [ 9 , 10 , 14 ]. DiRECT has clearly demonstrated that remission of T2DM is feasible and durable by dietary weight loss in the routine primary care environment [ 11 , 12 ]. In addition, the mechanistic studies of DiRECT confirmed and extended our previous observations related to T2DM development. It has been confirmed that a decrease in both hepatic and intrapancreatic fat is a prerequisite for diabetes remission provided that β-cell ability to recover after removal of this metabolic burden is retained [ 15 ]. Recently, the central importance of hepatic lipoprotein export on intrapancreatic fat accumulation and β-cell function was shown to be associated with both remission and redevelopment of T2DM [ 16 ]. Moreover, re-emergence of diabetes was related to increased enrichment of palmitic acid within the lipoproteins exported from the liver. This is important as palmitic acid is the obligatory product of de-novo lipogenesis (DNL) and the most toxic fatty acid to the β-cell. Further work is needed to understand the disordered lipoprotein and lipid metabolism in T2DM, especially in view of the increased risk of cardiovascular diseases. Many questions remain to be answered about lipid species, and fatty acid intermediates associated T2DM development and their relation to cardiovascular risks [ 17 , 18 ].
In this review, the pathogenesis of T2DM will be discussed from the perspective of the twin cycle hypothesis and its related studies. The disordered lipid metabolism related to the change in hepatic lipoprotein metabolism will be explained. Specifically, the effect of toxic lipid metabolites on β-cell function in respect to both diabetes development and remission will be discussed. Furthermore, our observations about abnormal pancreas morphology in T2DM and potential effects on the pathogenesis of this disease will be highlighted.
The twin cycle
It is over a decade since the twin cycle hypothesis, describing the route for reversibility of T2DM, was postulated [ 13 ]. It hypothesized that excess calorie intake over long term will divert excess energy storage towards the liver and other ectopic sites in the form of triglycerides. Excess lipid in the liver will decrease hepatocytes response to insulin leading to hepatic insulin resistance, thereby failing to switch off gluconeogenesis resulting in high plasma glucose and subsequently insulin levels. It was demonstrated that de-novo synthesis of fatty acids contributes largely to hepatic steatosis in human and animal models, and this is largely stimulated by insulin [ 19 , 20 ]. In T2DM, this will initiate the vicious cycle of hyperlipidaemia and hyperglycaemia due to a high basal insulin level. In the early years during T2DM development, β-cells respond to hepatic insulin resistance by increasing insulin secretion, raising the basal insulin level and reinforcing the liver cycle. Under these circumstances, hepatic export of very low-density lipoprotein triglycerides (VLDL-TG) will increase, pushing up the triglyceride level in circulation [ 21 ]. Subcutaneous adipose tissue provides a metabolically well tolerated fat storage area, but its capacity for storage is limited to a different extent in different individuals. In the face of increased hepatic VLDL-TG export, a personal fat threshold will be exceeded and ectopic fat accumulation will occur within the pancreas and other tissues [ 22 ]. This will initiate the pancreas cycle, whereby toxic fat metabolites will cause β-cell dysfunction in susceptible individuals. This is tolerated at the early stage of disease progress due to the compensatory ability of β-cell. However, when β-cell fail to compensate for increased loss of their functional mass, T2DM will emerge (Fig. (Fig.1 1 ).

The twin cycle hypothesis of the aetiology of T2DM. Liver cycle: Prolonged exposure of excess calorie intake under pre-existed muscle insulin resistance will divert energy storage from glycogen storage into adipose tissues through activating de-novo lipogenesis (DNL) pathway. When the maximum subcutaneous fat storage capacity is reached or fat storage within the adipocytes is impaired, the plasma triglycerides level will rise and diverted into the liver. Toxic lipid intermediates from triglycerides and fatty acids metabolism will cause hepatic insulin resistance, which leads to elevated levels of fasting insulin to compensate for insulin resistance. High insulin levels will enhance DNL and enforce the liver cycle. Pancreas cycle: Excess fat accumulation within the liver leads to elevation in hepatic VLDL-TG export to other tissues. This will increase exposure of the pancreas to high triglyceride concentrations, which increase fatty acids uptake and storage within the pancreatic tissues initiating the pancreas cycle. A long-term exposure of fatty acids and related toxic metabolites including high glucose would lead to impairment in β-cell function. β-cell will overcome this stress in early years during diabetes onset by secreting more insulin. However, when 50–60% of β-cell became dysfunctional, β-cell fail to maintain normal blood glucose and T2DM will emerge. High glycaemia associated with high fasting insulinaemia will drive more DNL which will enforce both the liver and pancreas cycles. Adapted with permission from [ 13 ]. T2DM, type 2 diabetes; VLDL-TG, very low-density lipoprotein triglycerides.
Liver fat and insulin resistance
The liver is central to regulation of blood glucose through endogenous glucose production. The association between insulin resistance and nonalcoholic fatty liver disease (NAFLD) is well documented [ 23 – 26 ]. Although it is not clear whether NAFLD is a causative factor or a result of insulin resistance, it is widely accepted that both are related to the pathogenesis of T2DM [ 24 , 27 , 28 ]. Elucidation of the exact mechanism of hepatic insulin resistance has been a major focus for many research groups [ 27 , 29 , 30 ]. Recent data from animal and human studies highlighted the role of diacylglycerol (DAG) in activation of hepatic protein kinase Cε (PKCε), which impairs insulin signalling [ 31 – 33 ]. In addition, saturated fatty acids also activate toll-like receptor 4 (TLR-4) in the liver and generate ceramides potentially inhibiting insulin signalling [ 33 – 35 ]. However, there are contradicting reports about the role of ceramides in driving insulin resistance in human [ 36 , 37 ].
NAFLD is known to increase cardiovascular risks in patients with T2DM [ 28 ]. This is likely to be related to a high atherogenic profile associated with altered lipid metabolism. In our studies, almost all people with T2DM have NAFLD at varied degrees, and liver fat was normalized rapidly after weight loss, Fig. Fig.2 2 [ 9 , 10 , 15 ]. Importantly, this was associated with major decrease in hepatic VLDL-TG export and normalization of hepatic insulin resistance, Fig. Fig.2 2 [ 10 , 16 ].

Change in liver fat, hepatic insulin resistance, and VLDL-TG production within the Counterbalance study. Hepatic triglyceride content (a), hepatic insulin resistance index (b), and hepatic VLDL1-triglyceride production (c) in those who reversed diabetes (responders: fasting blood glucose <7 mmol/L after return to isocaloric diet) and in those who failed to achieve reverse (nonresponders) at baseline (hatched bars), after VLCD (checkered bars), and after 6 months of weight maintenance (striped bars). * P < 0.05 for baseline–to–post-VLCD difference; # P < 0.05 for baseline–to–month 6 difference. Taken from [ 10 ], with permission from the American Diabetes Association. IR, insulin resistance; T2DM, type 2 diabetes; VLCD: very low calorie diet; VLDL-TG, very low-density lipoprotein triglycerides.
Polymorphisms in several genes are related to NAFLD; the PNPLA3 gene was reported to be strongly associated with NAFLD [ 38 ]. Work is currently ongoing to analyse the effect PNPLA3 polymorphism on lipid metabolism and T2DM remission within DiRECT.
Hepatic triglycerides export and lipoprotein metabolism
Another major function of the liver is to maintain lipid homeostasis. This is mainly regulated through VLDL-TG export and clearance of other lipoprotein remnants. In T2DM, lipid metabolism is abnormal, and this is a major risk factor for cardiovascular disease (CVD) development [ 17 , 18 ]. Disordered lipid metabolism in T2DM is characterized by overproduction of hepatic VLDL-TG [ 21 ]. This in turn is related to the expression of transcription factors that activate lipogenesis genes under elevated levels of glucose and insulin [ 39 ]. Free fatty acids derived from adipose tissue lipolysis are the major substrate for VLDL-TG production under fasting condition in healthy individuals. However, in T2DM, the contribution of DNL rises substantially [ 40 , 41 ]. We reported a major fall in hepatic VLDL-TG production after weight loss and this was significant only in those who achieved remission of diabetes, Fig. Fig.3 3 [ 15 , 16 ]. This decrease was associated with sustained normalization of plasma concentration of plasma total and VLDL-specific triglycerides provided remission was maintained [ 16 ]. In those who did not achieve remission, the changes in both VLDL-TG production and plasma VLDL-TG concentration were modest (Fig. (Fig.3). 3 ). On the other hand, loss of remission was associated with major rise in hepatic VLDL-TG production and plasma VLDL-TG concentration (Fig. (Fig.4), 4 ), which suggest a causative effect of VLDL-TG on T2DM development although this work required to be tested in a suitable animal model to prove causality [ 16 ].

Change in lipid parameters after remission of T2DM within the DiRECT study. Liver fat (a), fasting plasma insulin (b), total plasma triglyceride (c), hepatic VLDL1-TG production (d), fasting plasma VLDL1-TG (e), and VLDL1-TG pool (f) at baseline, post weight loss (5, 12, and 24 months). Responders are presented as a solid black line, nonresponders as a solid grey line, and nondiabetic controls (NDC: measured on one occasion) as a dotted line. Data are presented as means ± SEM. Weight loss itself brought about no significant differences between responders and nonresponders at any time point. Responders vs. baseline: * P < 0.05, ** P < 0.01, *** P < 0.001. Nonresponders vs. baseline: † P < 0.05, †† P < 0.01, ††† P < 0.001. Responders vs. 5 months: ‡ P < 0.05, ‡‡ P < 0.01. Nonresponders vs. 5 months: # P < 0.05, ## P < 0.01; Responders vs. NDC: ‡ P < 0.05, ‡‡ P < 0.01, ‡‡‡ P < 0.001; Nonresponders vs. NDC: + P < 0.05, ++ P < 0.01, +++ P < 0.001. Responders vs. nonresponders: ¥¥ P < 0.001. Reproduced from [ 16 ] with permission from Cell Metabolism. DiRECT, Diabetes Remission Clinical Trial; T2DM, type 2 diabetes; VLDL-TG, very low-density lipoprotein triglycerides. Responders: Those who achieved remission of diabetes: HbA1c<48 mmol/mol (6.5%) and fasting blood glucose <7.0 mmol/l off all anti-diabetes medication.

β-cell failure in response to change in lipid parameters during re-emergence of T2DM within DiRECT study. Change from baseline in fasting plasma glucose (a), fasting plasma insulin (b), liver fat (c), hepatic VLDL1-TG production (d), fasting plasma VLDL1-TG (e), total plasma triglyceride (f), intrapancreatic fat (g), and β-cell function (h) at 5 months (responders n = 38; relapsers n = 13), 12 months ( n = 28/ n = 13, respectively), and 24 months ( n = 20/ n = 13, respectively). Responders are presented as a solid black line and relapsers as a dashed line. The dotted line is the gridline at y value = 0. Paired data between baseline and each time point are presented. Data are presented as mean ± SEM except for the first phase insulin (median with IQ range) vs. 5 months in relapsers: * P < 0.05, ** P < 0.01, *** P < 0.001. Taken from [ 16 ], with permission from Cell Metabolism. DiRECT, Diabetes Remission Clinical Trial; T2DM, type 2 diabetes; VLDL-TG, very low-density lipoprotein triglycerides. Responders: Those who achieved remission of diabetes: HbA1c<48 mmol/mol (6.5%) and fasting blood glucose <7.0 mmol/l off all anti-diabetes medicationRelapsers: Those who returned to diabetes state after initial remission.
ApoB and ApoE are two major lipoproteins that regulate VLDL secretion and metabolism. Liver synthesis of ApoB is essential for successful assembly and secretion of VLDL particles, and this could be a regulatory process [ 42 ]. In contract, ApoE determines the hepatic uptake of lipoprotein remnants through binding to specific receptors on the hepatocyte. ApoE is therefore critical for clearance of these highly atherogenic lipoprotein species from circulation [ 43 ]. We reported that the HDL cholesterol level increased significantly after remission of T2DM [ 16 ]. It would be therefore of interest to study the change in plasma ApoB and ApoE kinetics following remission of diabetes. Moreover, genetic polymorphisms in the ApoE gens were reported to affect lipid metabolism in Alzheimer and CVD [ 44 , 45 ]. The ApoE genotyping study in respect to the pathogenesis of T2DM would be of major relevance to understand lipoprotein and lipid metabolism disorders in T2DM, and this is currently underway for DiRECT study.
Adipose tissue storage
Energy from excess calorie intake has to be stored for future usage. Glycogen synthesis allows storage of glucose in the liver and muscle, and this process is regulated by insulin function [ 46 ]. When glycogen stores are filled, excess energy will be diverted as triglycerides into adipocytes, normally located in subcutaneous adipose tissues under the skin. In T2DM, hepatic and muscle insulin resistance will limit glycogen storage and will drive excess energy towards the adipose tissues [ 47 ]. Indeed, high levels of glucose and insulin can stimulate transcription factors of lipogenesis, which make this pathway dominant in T2DM [ 39 ].
The capacity of subcutaneous adipose storage is limited, and this is determined by several factors including genetics, sex, and age [ 48 – 50 ]. In fact, these are the major factors that determine susceptibility to develop T2DM among individuals [ 22 ]. Moreover, inflammation was reported to suppress adipose tissue capacity for expansion [ 51 , 52 ]. There are several alterations in metabolic processes associated with expansion of adipose tissues in T2DM. First, insulin function in suppression of lipolysis would be limited due to insulin resistance in adipose tissues [ 53 , 54 ]. As a result, hepatic VLDL-TG production will rise due to high fatty acids substrate coming from lipolysis of the adipose tissue. Second, excess energy in the form of VLDL-TG will be diverted into circulation to be saved into and around internal body organs [ 13 , 21 ]. Third, high concentrations of saturated fatty acids can elicit inflammatory response [ 55 ], which can downregulate the capacity of adipose storage and directing fat to ectopic sites [ 51 ]. Finally, metabolism of excess triglycerides within the liver and pancreas will result in toxic lipid metabolites activating cellular process that impairs both hepatocyte and β-cell function [ 14 , 15 ].
In addition to store energy, adipocyte produces several regulatory adipokines that can affect our metabolism [ 55 ]. Leptin and adiponectin were reported to have antidiabetic effects through regulation of glucose and fatty acids metabolism [ 56 – 58 ]. Moreover, the plasma-leptin-to-adiponectin ratio is considered a marker of atherogenicity in T2DM and metabolic syndrome [ 59 , 60 ]. In this review, white adipose tissues that compromise the majority of the total body adipose tissues were discussed. However, brown adipose tissues, which are beyond the scope of this review, have important regulatory function on body metabolism [ 61 ]. Understanding the biology of these brown adipose tissues and their role in T2DM pathogenesis may permit greater understanding.
Pancreas fat and β-cell function
The concept of fatty pancreas is becoming widely accepted, and this has been reported to be common in most pancreas-related diseases including T2DM, pancreatitis, and pancreatic cancers [ 15 , 62 , 63 ]. We reported that intrapancreatic fat is elevated in people with T2DM compared with nondiabetic controls and reversal of diabetes has always been associated with a major decrease in pancreas fat and normalization of insulin secretion [ 9 , 10 , 15 , 16 , 64 , 65 ]. According to the twin cycle hypothesis, excess fat is delivered to the pancreas via hepatic VLDL-TG export [ 13 ], and initial studies demonstrated that fall in VLDL-TG export was associated with a gradual decrease in intrapancreatic fat concurrent with the restoration of β-cell function [ 9 , 10 ]. Further studies in DiRECT confirmed those findings, and showed that changes in VLDL-TG production and intrapancreatic fat content to be related to both diabetes reversal and redevelopment, Fig. 4 [ 15 , 16 ]. Furthermore, we have confirmed that a specific enrichment of palmitic acid within the VLDL-TG is likely to drive these processes [ 16 ]. The deleterious effect of saturated fatty acids on β-cell function has been known for a long time [ 66 ], and several concepts to explain the lipid-induced β-cell damage were proposed including cell apoptosis and cell dedifferentiation [ 6 , 67 , 68 ]. The exact mechanism that causes β-cell dysfunction remains uncertain [ 6 , 7 , 69 – 71 ]. However, β-cell dedifferentiation appears to be the most likely and has become the most widely accepted to explain β-cell failure in T2DM [ 67 , 72 , 73 ]. It proposes that under metabolic conditions of excess fat and eventually glucose, β-cell is converted to an α-cell phenotype [ 73 , 74 ]. Conclusive data about β-cell dedifferentiation are limited, especially in human studies [ 73 , 75 ]. Recently, two major regulators of β-cell dedifferentiation were reported [ 76 ]. Considering the lack of β-cell–specific markers, identification of generic biomarkers for cell stress and differentiation would be useful. In this respect, growth and differentiation factor-15 (GDF-15) and fibroblast growth factor-21(FGF-21) could serve a potential candidates considering reports of their effect on lipid metabolism and nutritional stress in T2DM [ 77 – 79 ]. More work is needed to exclusively determine the factor (s) that cause β-cell dysfunction and recovery in T2DM.
Data from animal and human studies indicate the potential role of saturated fatty acids in inducing endoplasmic reticulum (ER) stress that lead to β-cell dysfunction [ 80 – 83 ]. Recent studies emphasized the role of branched-chain amino acids (BCAAs) in T2DM [ 84 – 86 ]. It has been reported that BCAA stimulates insulin secretion and activates the mTORC1 kinase which is related to β-cell mass and function [ 87 ]. mTORC1 is a negative regulator of autophagy, a process that known to regulate lipid metabolism [ 88 ], is reported to be regulated by calorie restriction [ 89 , 90 ]. β-cell autophagy was reported to be abnormal under condition of high lipids, and removing this metabolic stress restored autophagy function [ 91 ]. Several cellular mechanisms were proposed to explain β-cell dedifferentiation [ 75 ]. Clinical and metabolic studies together with other cellular and animal studies support that the process is driven by fat-induced metabolites that cause ER stress [ 72 , 81 , 82 ] leading to β-cell dysfunction. Our data confirm that β-cell damage is reversible in the early years after diabetes onset, which supports the theory of β-cell dedifferentiation rather than apoptosis. The process underlying reversal of T2DM is likely to be redifferentiation following removal of the toxic metabolic conditions after weight loss, but this remains to be determined experimentally [ 14 ]. It is possible that normalization of autophagy function after decreasing β-cell exposure to palmitic acid might contribute to β-cell redifferentiation.
Pancreas morphology
Despite its central importance to whole body metabolism, the pancreas remains one of the least studied organs. This is largely due to the complex anatomical structural and deep position within the abdomen [ 92 ]. In the past 20 years, magnetic resonance techniques have emerged as one of the most useful tools to study this organ [ 93 ]. We have successfully developed and employed techniques to study pancreas morphology and fat content in T2DM [ 9 , 65 , 94 , 95 ]. We found that pancreas volume is around one-third lower than normal in T2DM and the organ has very irregular borders, present soon after diagnosis and the volume appears to decrease further with increasing duration of diabetes [ 94 , 95 ]. It has not been established whether T2DM develops more readily in those born with a small pancreas or loss of volume is secondary to the disease process. Insulin acts as a potent growth hormone at high concentration such as experienced by parenchymal pancreas tissues by paracrine action after a meal [ 47 , 96 ]. Therefore, lack of local acute insulin secretion in T2DM may explain the decline in pancreas volume. In support of this notion, it was reported that pancreas volume is decreased in people diagnosed with type 1 diabetes, where local insulin secretion is completely absent [ 97 , 98 ]. Restoration of insulin secretion did not bring about any improvement in pancreas volume during the first 6 months after diabetes remission [ 95 ]. However, this is expected considering the long-term of insulin deficiency experienced by pancreatic tissues during T2DM onset, which is expected to be around 10 years, and a longer-term fellow-up within DiRECT revealed a significant increase in pancreas volume only in those who achieved return of insulin secretion [ 99 ]. It is notable that insulin and insulin like growth factor-1 (IGF-1) receptors share high homology, and insulin can therefore bind to the IGF-1 receptor at low affinity, which explain the trophic effects of insulin at high concentration [ 100 ]. It is possible that lack of both IGF-1 and insulin are involved in pancreatic tissue atrophy observed to happen in T2DM. Further work on the change in IGF-1 levels following T2DM remission, and how this could be related to change in pancreas volume is required.
Synthesis of information
The twin cycle hypothesis and evolving studies have changed the perception about T2DM being a long-term progressive disease. The counterpoint study demonstrated remission from T2DM for the first time, and the counterbalance identified the effect of diabetes duration on the likelihood of return to normal blood control [ 9 , 10 ]. Recently, DiRECT extended our previous findings [ 11 , 12 ]. This had changed the clinical guidelines, which now acknowledge and apply definitive weight loss in T2DM management programmes [ 101 ]. Understanding the pathophysiological processes that lead to T2DM development and reversal is crucial to control this disease. The twin cycle hypothesis was formulated after prior study of liver metabolism. It outlined the main aetiological features of T2DM and predicted the potential route for reversal [ 13 , 102 ]. Until now, the exact mechanism(s) that explain how T2DM is reversed after weight loss are lacking, but hepatic insulin resistance and β-cell dysfunction have now been shown to be the major determining factors for the pathogenesis of this disease [ 14 ]. There is an accumulated body of evidence from clinical and metabolic studies over the past 10-15 years [ 9 , 10 , 15 , 16 ] supported by other cellular and animal studies, all emphasized the deleterious effect of lipids on hepatic insulin resistance [ 29 , 32 , 33 , 103 ] and β-cell function [ 80 – 83 ].
The lipotoxic effect of fat-derived metabolites on the hepatic insulin resistance is widely accepted. Toxic lipid intermediates were derived from fatty acids metabolism including DAG and ceramides [ 36 , 103 ]. In the pancreas, there are conflicting opinions whether lipotoxicity or glucotoxicty is the cause of β-cell dysfunction in T2DM [ 69 ]. It is difficult to rule out the effect of glucose from the effect of fatty acids. However, it is clear that high glucose levels cannot initiate the process. Once T2DM is established, it is likely that both glucose and fatty acids could synergistically add to the metabolic stress and lead to β-cell dysfunction [ 70 , 104 ], and this is more likely at advanced stages during the progress of this disease. However, there has to be an initiating factor for loss of β-cell capacity before diabetes onset. The precise mechanism of fat/glucose-induced damage to the β-cell is not finally established. Apoptosis or cell death can be observed after in-vitro β-cell exposure to saturated fatty acids [ 66 , 69 , 105 – 107 ]. However, it has been demonstrated recently that β-cell undergoes dedifferentiation rather than apoptosis under metabolic stress [ 67 , 73 , 75 , 108 , 109 ]. Data derived from diabetes reversal after weight loss support the dedifferentiation concept and propose that the return of β-cell function can happen through the process of redifferentiation [ 72 ].
One of the major predictions of the twin cycle hypothesis is hepatic VLDL-TG being the upstream process to deliver the toxic lipid metabolites to peripheral tissues including the pancreas [ 13 ]. We and others have confirmed that hepatic VLDL-TG production is elevated in T2DM [ 10 , 13 , 15 , 21 ]. We also found that the change in hepatic VLDL-TG production was associated with both remission and redevelopment of T2DM [ 10 , 15 , 16 ]. It has been established for many years that prolonged β-cell exposure to saturated fatty acids is harmful [ 81 , 105 , 110 ]. Palmitic acid is predominately produced during DNL, and incubation of β-cells with relatively low concentration of palmitic acid induced ER stress [ 81 , 83 , 107 ]. In obesity and T2DM, palmitic acid was reported to initiate the inflammatory response that causes β-cell damage via TLR4-dependent pathway [ 110 , 111 ]. Furthermore, T2DM is associated with raised levels of inflammatory cytokines including interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), and nuclear factor kappa-B (NF-κB) [ 107 , 112 , 113 ]. Importantly, the palmitic acid content of the exported VLDL-TG was shown to decrease after remission and markedly increase in response to relapse into diabetes [ 16 ]. Taken together, these data suggest the important role of hepatic VLDL-TG in delivering toxic lipid metabolites that may initiate harmful cellular process. Further work is required to verify the causality factor of VLDL-TG on β-cell function.
The pancreas is composed of endocrine and exocrine systems, and both systems are required to maintain normal function. We have reported that pancreas morphology is abnormal in T2DM, and this was evident from the time of diagnosis [ 94 , 95 ]. Acinar cell mass reflects the total pancreas volume considering the small contribution of islet and ductal systems (~5%). Clinical and observational studies of the pancreas have naturally focussed on islet function itself, but the relevance of acinar cells to endocrine function has not been considered [ 114 – 117 ]. While intrapancreatic fat was found to increase with long diabetes duration, both β-cell function and pancreas volume were reported to decline [ 10 , 95 , 118 ] raising the question of possible causative relationships between these variables. In the ZDF rat model of T2DM, fat replacement of the acinar cells developed into fibrosis [ 119 ], and advanced fibrosis may lead to destruction of the islets and β-cell dysfunction [ 116 , 119 – 121 ]. Immunohistochemistry studies of postmortem tissues of pancreas from people with T2DM indicated that loss of β-cell function was associated with acinar cell fibrosis [ 122 ]. Plasticity and regeneration of acinar cells are well documented [ 123 – 125 ], and elucidation of the physiological and molecular associations of acinar cell mass regeneration is required. IGF-1 is anabolic and growth factor hormone produced mainly in the liver [ 126 ]. If the trophic effect of insulin affects pancreas volume, IGF-1 would be expected to have a greater effect due to its higher growth function ability. This is likely to be achieved in collaboration with insulin considering the affinity between insulin and IGF-1 binding protein [ 100 ]. IGF-1 has been reported to decrease in type 1 diabetes, ageing, and T2DM [ 127 – 130 ] where pancreas volume was reported to decline [ 94 , 97 , 118 ]. Additionally, studies on postmortem pancreas of people with T2DM have shown fibrosis in exocrine tissues associated with decline in β-cell and increase in α-cell mass [ 122 ]. Whether this morphometric changes in acinar cells are related or secondary to loss of β-cell function warrants more investigation.
In summary, T2DM can now be viewed as a state of excess liver and intrapancreatic fat content. The underlying pathophysiologic mechanisms that determine diabetes development and remission are partially understood. However, disordered hepatic VLDL-TG export and the associated abnormalities in lipid metabolism during excess calorie intake appear to be central to the pathogenesis of this disease. Further investigations to identify toxic lipid metabolites and the precise in-vivo mechanisms by which they lead to β-cell dysfunction are required. Heterogeneity of T2DM has been overstated and relates largely to individual capacities both for subcutaneous storage of fat and susceptibility to fat-induced β-cell dysfunction. There is major overlap in the basic pathogenesis of both pathogenesis of both CVD and T2DM. Additionally, the relevance of abnormal pancreas morphology to the pathogenesis of T2DM requires more definitive study to identify the factors that cause acinar cell loss and regeneration and how this may affect β-cell function.
Acknowledgements
I would like to acknowledge Professor Roy Taylor and all members of his wider research team over the past 12 years involved in the Counterpoint, Counterbalance, and DiRECT studies. Diabetes UK funded Counterpoint and DiRECT. Counterbalance was funded by Newcastle Biomedical Research Centre and supported by a fellowship from the Novo Nordisk Foundation.
Conflicts of interest
There is no conflict of interest.
IMAGES
VIDEO
COMMENTS
Transcription overview. Transcription is the first step of gene expression. During this process, the DNA sequence of a gene is copied into RNA. Before transcription can take place, the DNA double helix must unwind near the gene that is getting transcribed. The region of opened-up DNA is called a transcription bubble.
The transcription factor cycle (TFC) hypothesis suggests the transfer of DNA-bound factors to nascent RNA. Exerting downstream functions in RNA processing and transport, these factors would be liberated by RNA processing and cycle back to the DNA maintaining active transcription. Sequestered on RNA in absence of processing they would constitute ...
Moreover, each potential hypothesis is not mutually exclusive and may be true for only a subset of cell cycle-regulated genes. ... enable the construction of global transcription factor networks that describe the regulatory interactions of the cell cycle transcription program [8, 39, 43, 44, 54, 72, 73].
The typical RNA polymerase II (Pol II) transcription cycle begins with the binding of activators upstream of the core promoter (including the TATA box and transcription start site). This event leads to the recruitment of the adaptor complexes such as SAGA ... This hypothesis is supported by the observation that in a paf1Δ mutant, both H3K4me2 ...
Comparative genome-wide analyses reveal that DNA replication origins fire near the initiation site of highly transcribed genes, ensuring co-directional replication and transcription in highly ...
Together these findings demonstrate that, contrary to the 'transcription cycle' hypothesis, TLs of long highly expressed genes are open-ended loops with separated flanks. TLs are larger for ...
Significance. Our study provided a comprehensive view of the transcriptional landscape across the cell cycle. We revealed lag between transcription and steady-state RNA expression at the cell-cycle level and characterized a large amount of active transcription during early mitosis. In addition, our analysis identified thousands of enhancer RNAs ...
The Transcription Factor cycle hypothesis: The basic propositions are given in the text. In short : (1) TFs (and other DNA-binding factors) attach to oligomotifs in the DNA at Cis-Regulatory ...
During the first stage of the transcription cycle, ... We corrected for multiple hypothesis testing using the false discovery rate (Storey and Tibshirani, 2003) and filtered ASTSC shapes using a 10% FDR. Applying the same criteria and K-S test to biological replicates (mice F5 and F6), identified fewer candidate differences between biological ...
transcription, the synthesis of RNA from DNA.Genetic information flows from DNA into protein, the substance that gives an organism its form.This flow of information occurs through the sequential processes of transcription (DNA to RNA) and translation (RNA to protein). Transcription occurs when there is a need for a particular gene product at a specific time or in a specific tissue.
Two conserved processes express the genetic information of all organisms. First, DNA is transcribed into a messenger RNA (mRNA) by the multisubunit enzyme RNA polymerase (RNAP). Second, the mRNA directs protein synthesis, when the ribosome translates its nucleotide sequence to amino acids using the genetic code.
Our theoretical model and agreement with general trends in scRNA-seq data supports the hypothesis that a cell cycle-dependent transcriptional filter has the potential to control cell proportion and diversity in tissue development. ... We must also more carefully consider cell cycle phase as transcription mainly occurs in the gap phases (Bertoli ...
The transcription factor cycle (TFC) hypothesis suggests the transfer of DNA-bound factors to nascent RNA. Exerting downstream functions in RNA processing and transport, these factors would be liberated by RNA processing and cycle back to the DNA maintaining active transcription. Sequestered on RNA in absence of processing they would constitute ...
One theory about how the different mRNA-making processes are coordinated is called kinetic competition. This theory states that the fastest process is the most likely to occur, even if the other processes use less energy and so might be expected to be preferred. ... Kinetic competition during the transcription cycle has been shown to influence ...
Explain the role of RNA polymerase during transcription., b) Identify the dependent variable in the experiments. ... rubiscofixes atmospheric carbon into an organic molecule as one of the first steps in the Calvin cycle. ... Researchers design an experiment to test the hypothesis that the evolution of coloration in lice is driven by birds ...
Chapter 12&13 Homework. You are studying a transcription factor that controls entry into the cell cycle. It is present in the nucleus of nondividing cells, but rapidly exits the nucleus when cells are stimulated to begin cell division. You hypothesize that the transcription factor represses expression of genes that drive entry into the cell ...
This theory states that the fastest process is the most likely to occur, even if the other processes use less energy and so might be expected to be preferred. ... Kinetic competition during the transcription cycle has been shown to influence splice site selection during alternative splicing (de la Mata et al., 2003), recruitment of factors ...
Particularly, if you find yourself in Krasnodar, we recommend you to visit the Krasnodar Regional Art Museum named after F. A. Kovalenko to enjoy a rare collection of Russian avant-garde and Dutch art of XVI century, and also attend concerts of SSAI "Kuban Cossack chorus" to listen to Kuban Cossack, Russian and Ukrainian folk-songs.
The twin cycle hypothesis postulated that both hepatic insulin resistance and dysfunction rather than death of beta (β) cell determine diabetes onset. ... This in turn is related to the expression of transcription factors that activate lipogenesis genes under elevated levels of glucose and insulin . Free fatty acids derived from adipose tissue ...
نادي: كراسنودار - كووورة ... نادي: كراسنودار
The Project provides for the construction of a new combined cycle gas turbine unit (CCGT unit) with an electric capacity of 410 MW and a heat power production of 220 Gcal/h. Natural gas is used as prime and reserve fuel; diesel fuel is used in the event of emergencies. The new CCGT unit (TGK-410) will be located in a new building being constructed
facility at the Krasnodar CHP Plant, i.e. a 410 MW combined cycle gas turbine (CCGT) unit. A new building for the unit will be constructed in the northern part of the existing Plant's site. A detailed technical and environmental design of the project is being undertaken by a Russian general engineering and procurement contractor "E4".