Data Management
- Quality Assurance
- Trial Sites
- Clinical Pharmacy
- Participate in a Study
- Task Research Academy
- Community News

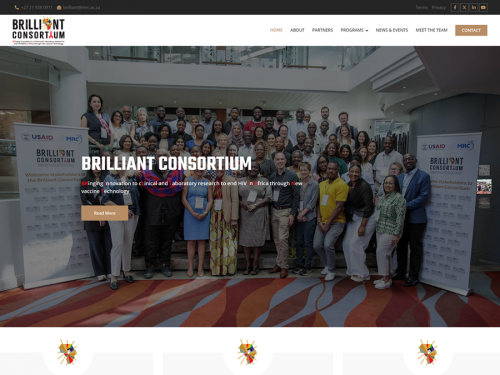
TASK is a multinational, multi-site clinical research institute committed to improving global health through testing and progressing novelty medicines, vaccines and diagnostics in various therapeutic areas.

Our Network of Sites
- We have 7 sites providing expertly managed clinical studies to patients
- 19+ years of experience working with sponsors and CROs conducting hundreds of clinical trials in many indications
Clinical Trials
Regulatory Services
BSL-3 laboratory
International Accredited Training
Get in touch
Smith Street, Glenlily, Cape Town,7500
+27 (0)21 100 3606

" * " indicates required fields
Copyright © 2023 TASK – All Rights Reserved. | Privacy Policy | Resources | Communication Policy | Briefs

FACILITATE | EDUCATE | MENTOR | ADVOCATE

Positioning South Africa at the Forefront of Clinical Research
Stay updated with the latest information at www.sacoronavirus.co.za

About SACRA
A Distinct Identity
The South African Clinical research association (SACRA) is an industry community association with the sole purpose of leading and serving as a conduit within the clinical trials community. SACRA is a non-profit organisation representing the clinical research industry in South Africa.
SACRA's Purpose
To lead and serve as a conduit across the various stakeholders, positioning the country at the forefront of clinical research both nationally and internationally.
SACRA's Values

SCIENTIFIC-RIGOUR

INCLUSIVITY

Latest News
We work hard to get our efforts noticed by the media and are so proud when this goal comes to fruition. A well informed community is an empowered one as well, so take a look at some of the latest coverage we’ve received below and help spread the word about all the amazing developments at SACRA.

SACRA AGM 10 April 2024 - Presentations published

Upcoming Programme: Clinical Research Education Program

Workshop: Diversity, Equity and Inclusion in Healthcare (DEI)
Upcoming Events

We love hearing from SACRA stakeholders
If you have any news or comments, send them to us now!
Please send your comments and suggestions to
Thanks for submitting!
The SAMRC was established in 1969 and is dedicated to improving the health of people in South Africa, through research, innovation, development, and technology transfer. The scope of research includes laboratory investigations, clinical research, and public health studies.
We conduct research on South Africa’s quadruple burden of disease: maternal, newborn and child health, HIV/AIDS and TB, non-communicable diseases, and interpersonal violence. Our work is to acquire evidence-based information to inform health policy and practice and improve the quality and health status of people in South Africa.
Vision & Mission
Building a healthy nation through research, innovation and transformation
To advance the nation’s health and quality of life and address inequity by conducting and funding relevant and responsive health research, capacity development, innovation and research translation
Latest News

Latest Resource Materials

SAMRC Project/Collaboration Websites
Event Calendar

Our Services

Biostatistical Analysis

Conference and Event Planning

Genomics Platform

Health Geographic Information Systems

Knowledge and Information Management Services

Research and Innovation Management System
Covid-19 | Subscribe
Clinical Trials
The Clinical Trial Unit of the South African Health Products Regulatory Authority (SAHPRA) provides legal framework for the review of clinical trials and Bioequivalence studies for human participants and recommends approval of the conduct of clinical trials. The unit receives, processes and evaluates the applications from applicants (industry, academia and investigators) for approval to conduct the study within South Africa (SA). The unit also provide for authorisation for the importation of unregistered medicine for the purpose of conducting clinical trials. Any amendments required during the conduct of the study, must be approved by SAHPRA.

Legislative Framework
Medicines and Related Substance Act, Act 101 of 1965, provides for the legislative framework for access to unregistered medicines, it is enabled by Section 21 of the Act as amended: Authority may authorize sale of unregistered medicines for purpose of conducting clinical trials.
South African Good Clinical Practice Guidelines (SA GCP) provide researchers and other interested parties with clearly articulated standards of GCP in locally conducted research that address the local realities and contexts, to ensure that clinical trials involving South African human participants are designed and conducted according to local requirements as well as according to the sound scientific and ethical standards within the accepted framework for good clinical practice.
SAPRA has developed several guidelines for researchers in order to ensure that human participants are protected and are able to derive benefits from participating in clinical trials conducted in South Africa.
Processing Clinical Trials Applications
- Researchers must submit a completed application on predetermined dates and obtain proof of delivery. An application form must be accompanied by the prescribed fee. The proof of delivery, proof of payments and cover page must be sent to SAHPRA via email.
- SAHPRA New Clinical Trials Process
- Pre-Approval
- Post-Approval
- Processing of Application for Protocol Amendment and Additional Sites And/Investigators
SAHPRA New Clinical Trials Process / Pre-Approval

Turnaround Time for Response to a Clinical Trial Application
Clinical Trial Unit aim to process new applications and issue checklist within 3 weeks of receipt. The Clinical Trial Committee recommendations would be sent within 10 weeks of submission due date. There are cases where this turnaround time might be prolonged i.e. unfamiliar investigational product which may be referred to external reviewers or other committees of SAHPRA for input.
The timeline to receive the response after the submission of application for additional investigators, site(s) and protocol amendment is about 6 weeks following receipt of the application. The applicants should make use of the electronic submission process .
Fees Payable to SAHPRA
- Details of all clinical trials application whereby fees are applicable are available on: Fees payable to SAHPRA
- The South African National Clinical Trials Register (SANCTR) provides the public with updated information on clinical trials on human participants being conducted in South Africa. The Register provides you with information on a trials purpose; who can participate, where the trial is located, and contact details. Find out more .
- New clinical trials application alert, Responses to new Clinical Trial applications and related queries: [email protected]
- Protocol amendments, responses to amendments and related queries: [email protected]
- Additional Investigators & sites, responses to additional and related queries: [email protected]
- Bioequivalence studies, BE amendments, responses to BE studies and related queries: [email protected]
- Notifications and related queries: [email protected]
- Individual Patient Serious Adverse Events and related queries: [email protected]
- SAHPRA guidelines, forms and related queries: [email protected]

About CRISA
Clinical Research Institute of South Africa
Clinical Research Institute of South Africa (CRISA), Proprietary Limited. The purpose of CRISA is to conduct clinical research in South Africa in collaboration with Department of Health on projects that lead to optimised care for patients and promote a public health approach to research and treatment. CRISA aims to integrate clinical research into the clinical pathway in hospitals and clinics in iLembe Health District. more

Department Of Health
In KwaZulu Natal, most Clinical Research centres are situated in eThekwini District. To ensure equity of access to health support services, the KwaZulu Natal Department of Health and Clinical Research Institute of South Africa (CRISA) entered into a Memorandum of Understanding to govern the work of CRISA in iLembe.

Ilembe District Municipality
The iLembe District is situated on the East Coast of South Africa in the Province of KwaZulu Natal. It is in close proximity to the King Shaka International Airport and the Dube Trade Port. it is made up of four health sub-districts, namely Mandeni, KwaDukuza, Maphumulo and Ndwedwe.

KwaDukuza Minicipality
CRISA is situated at number 154 Goolam Suleman Street (Old Hulett Street) KwaDukuza Municipal area of jurisdiction, province of KwaZulu-Natal. Located 250m south of General Justice Ginzenga Mpanza Hospital (former Stanger Hospital)and approximately 800m north west of KwaDukuza Clinic. The site has accessibility and visibility to public transport route.

Operation Sukuma Sakhe
CRISA has an obligation to implement effective stakeholder engagement programme to build a platform of mutually beneficial, sustained relationships between trial sponsors, site and community where CRISA conducts clinical research. Through Operation Sukuma Sakhe (OSS), a diverse cultural, transparent and respectful platform is created to address interests of community stakeholders.
- SP_SOCIAL_INSTAGRAM
- Our History
- Global Footprint
- Our Partners
- Board of Directors
- Group Executive Management
- South Africa Executive Management
- Group Technical Leads
- Clinical Research
- Implementation Research
- Health Programmes
- Global Projects
- Aurum Institute Ghana
- Fundação Aurum
- Tuberculosis
- Health Systems
- Key Populations
- Antimicrobial resistance (AMR)
- Youth Health Africa
- Publications
- Annual Reports
- Annual Financials
- Latest News
- Press Releases
- Aurum in the News
- Image Gallery

Running clinical trials ranging from large scale public health studies, to highly regulated clinical trials of new medications.
Each clinical research site (crs) brings a unique complement of skills, populations, facilities, and experience., conducted > 210 trials and research studies in the past 18 years focusing on:, hiv vaccines & prevention (oral, topical, and long-acting injectable prep), tb treatment, host-directed therapy, tb vaccines & tb preventative therapy.
COVID-19 vaccines, COVID-19 mAb & COVID-19 treatment
Diagnostics and socio-behavioral studies., enrolled > 66 500 participants in trials and research studies (clinical research division), more than 23 500 thibela tb (enrollment at mine shafts), more than 25 500 sisonke vaccine roll-out to healthcare workers (enrollment at department of health vaccination sites), more than 17 500 enrolled in remaining trials and research studies (enrollment at 6 clinical research sites), aurum’s distinct strengths:, crs pluripotency, collective scientific, operational & management experience of its research leadership, high-functioning resources & systems to effectively implement large-scale complex clinical trials (both network & investigator-lead), we focus on these programmatic areas:.

HIV Treatment & PreP

TB Treatment
HIV Vaccine

Special Projects
2023 at a glance.
Aurum CRS Centres

We have seen the impact of clinical trials saving millions of lives in the COVID-19 pandemic and we are now even more committed to continue our search for safer, more effective tools to address the continuing TB and HIV pandemics!

Tanya Nielson Managing Director: Clinical Research Division

Dr Craig Innes Clinical Director: Clinical Research Division

Sharfuddin Sayed Operations Director: Clinical Research Division

Dr William Brumskine Clinical Research Site Leader: Rustenburg

Dr Kathy Mngadi Clinical Research Site Leader: Tembisa

Dr Vaneshree Govender Clinical Research Site Leader: Pretoria

Dr Pearl Selepe Clinical Research Site Leader: Klerksdorp

Trevor Beattie Technical Director: CRD Projects

Lindiwe Nhlangulela Head of Department: Laboratory

John Mdluli Head of Department: Community

Naydene Slabbert Head of Department: Data Management

Yajna Duki Head of Department - Quality Assurance
Latest Highlights

Aurum Becomes New Home for Key Assets from IAVI's Human Immunology Laboratory
Iavi’s human immunology laboratory relocates samples to africa, fulfilling capacity strengthening goal.

Advancements in HIV Prevention: A Conversation with Dr. Kathryn Mngadi

Beyond the Finish Line: The Aurum Klerksdorp Marathon

Aurum Leadership Roadshow takes Klerksdorp

Royal Bafokeng leader visits Aurum Rustenburg CRS
Health for mzansi, kzn’s dr cele reflects on the joy of his covid-19 milestone.

PanTB-HM Clinical Trial Begins Recruitment in South Africa
Pretoria north’s health and social outreach event hosted by metro, growing the beta variant – young scientist remembers the day they danced in the lab.

Key Takeaways from IAS 2023: Part 1

Aurum welcomes new TB funding commitment by donors
Clinical Research Division
Implementation Research Division
Health Programme
Aurum Global
Aurum Ghana
Aurum Mozambique
Head Office
29 Queens Road, Parktown, Johannesburg, South Africa, 2193. (+27) 10 590 1300 [email protected]
BBBEE Certificate | Disclaimer | Privacy Policy | PAIA | Information Access Request | Outcome of PAIA Request and Fees Payable | Research FCOI Policy | FCOI Disclosure Form
Welcome to OnQ
The african contract research organisation.
We are ideally placed to understand the nuances of conducting clinical research in African countries.
We're Committed to Helping You Run Successful Trials
Talk to us about doing clinical trials in africa, onq joins the p95 family, about onq sa, we are the african cro you can trust, we have been in the industry for over 20 years.
Established in 1999, OnQ Research has conducted more than 500 clinical trials in more than 10 African countries. Our clinical study sizes range from 1 to 3000 participants.
The OnQ Difference
We are dedicated to empowering the African continent through safe trials.
Catherine Lund
Our service offerings, we help you execute clinical trials successfully, project management services.
OnQ provides end-to-end project management solutions for the entire lifecyle of clinical trials
Mobile Nursing Services
Facilitating safe direct to patient services through home nursing.
Data Management Services
OnQ designs protocols and databases according to standard conventions.
Quality Assurance Services
OnQ collaborates with medical practitioners to ensure regulatory compliance throughout the clinical study lifecycle.
Medical Monitoring Services
OnQ has medical monitoring expertise from protocol design to clinical study report writing.
Vendor Management Services
Vendor mangement services.
OnQ centralises vendor management to simplify eco-system collaboration and study efficacy.
Pharmacovigilence Services
OnQ is fully committed to patient safety at all stages of clinical trials.
Medical Writing Services
OnQ supports medical practitioners to deliver regulatory-compliant clinical trial documents.
Site Management Services
OnQ performs site management services on site and remotely to ensure that the rights and well-being of study participants are protected.
Study Participants
Research sites, professionals, company achievement, popular service, take a look our unmatched consulting approach.
- Strategy & Innovation
- Quality Compliance
- Sales & Marketing
- Data Analytics
Management Consulting and Strategy Consulting
Sed ut perspiciatis unde omnis iste natus error sit voluptem accusantium doloremu laudantium totam rem aperiam eaque upsa quae abillo inventore veritatis architecto
- Static Innovations
- Consulting & Advisory
- Turnaround Situations
Meet Our Leadership Team
Managing Director & Founder
Teresa Scanes
System Operations Director
Nyeleti Rikhotso
Clinical Operations Director
Nyeleti Rickhotso
Work with us, meet our professional team.
Sed ut perspiciatis unde omnis iste natus error sit voluptam accusantium doloremque laudantium totam reme aperiam eaque quae inventore
Judson S. Gooden
Web Designer
Careers at OnQ
We are always looking for talent. send your cv, clinical research associates.
We are always on the lookout for talented Junior to Senior CRAs
- Any time, just share your cv
Project Managers
We offer all levels of opportunities for PM roles
- Anytime, just share your CV
Quality Assurance Specialists
Talented professionals with QA & Pharmacovigilance wanted
- Anytime, simply share your CV
Strong African Presence
We are in over 10 african countries, are you ready to work with us, request a call back, build a better career with us.
- [email protected]
- +27 11 431 0791
Site Management Organisation
WELCOME TO CRISMO
Clinical Research Investigator Site Management Organisation (CRISMO), a Site Management Organisation based in Germiston, South Africa.
Bridging the capacity building gap in Africa
Clinical Research Investigator Site Management Organisation (CRISMO), is a Site Management Organisation established in 2015 with its head-office based in Germiston, South Africa.
We bridge the capacity building gap in Africa by providing site management systems and operational support to research investigator sites in the conduct of clinical research and clinical trials through our world-class dedicate research sites (DRS) and affiliated sites.
Participants
Studies secured, affiliated sites, we innovative clinical trial execution strategies, protocol writing.
A brief outline of what the study is and how it is going to be carried out. Ensuring compliance with standards and regulatory requirements.
Patient Recruitment
Raise awareness of clinical trials and encourage participation on ongoing and future studies.
Data Analysis
Interpretation of data to derive a report and final study report
Data Collection
Gathering and measuring information on targeted variables in an established system which then enable one to answer relevant questions and evaluate outcomes
Our Latest News

Ensuring Patients Safety in Clinical Trials
Clinical trials play a crucial role in advancing medical science, driving innovation, and developing new treat ...
Laboratory specimens extracted during a clinical trial
Laboratory Specimen in clinical research refers to the sample of a participant’s tissue, fluids and any other ...

What is Data Management in Clinical Trials?
The primary goal of a clinical trial is to obtain valuable insights and answers to specific research questions ...
Our Partners and Stakeholders
Still have questions.
Feel free to go through our Frequently asked questions, and if you need clarity or more assistance do not hesitate to contact us..
Dreaming Of Joy
Click edit button to change this text. Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Crazy Circus
Accordion title, celebrity skin, feel free to ask something we are here.
Bertha Gxowa Hospital Ground Floor, Villa Heidi Building, Germiston, Gauteng Cnr. Joubert and Hospital Street
Phone: +27 11 038 6814 Email: [email protected]
Your message
Future Medicine Delivered Today
- Currently Enrolling Studies

The Limpopo Clinical Research Initiative has been at the forefront of making a difference in the healthcare delivery to our community since 2009. We support innovative thinkers who want to create a lasting change by addressing some of our greatest medical challenges.
We are delivering the data fundamental to the development of future medicines while investing international funding to improve local healthcare and tackle social challenges.
What We Want
We want to bring about the ultimate, global, specialised rural healthcare service of choice through clinical research excellence and value-added community outreach.

Capacity Building
The purpose of this Initiative is to do capacity building for clinical research in the previously under serviced geographical area of the Western Limpopo Province. This is done to add value to the health services of our local communities. Within the past 10 years, we have successfully introduced to clinical research and Good Clinical Practice [GCP] trained over 31 medical practitioners (23 Historically Disadvantaged Individuals [HDIs]), 17 registered nurses (3 HDIs), 3 enrolled nurses, 22 study coordinators (1 HDI), 5 pharmacists, 1 dietician, 1 nutritionist, 1 psychologist and 10 clinical trial assistants (9 HDIs). This includes GCP training that was provided to nursing personnel from the local public primary care clinics.

Home | Conducting Clinical Trials in South Africa: A Revitalized Environment
Conducting Clinical Trials in South Africa: A Revitalized Environment
South Africa was once a commonly considered country for inclusion in clinical research, but its popularity caused bottlenecks within its previously constructed regulatory structure. Simply put, the system became overwhelmed and could not keep up thus leading to long timelines, and therefore making the country less of an ideal candidate for inclusion. Recently, the South African government took action and made efforts to make the country a more supportive environment, so clinical research could return and at scale. They succeeded and we will discuss this in further detail.
Article Contents:
Previous bottlenecks in south african regulatory environment, process changes by the south african government, the new regulatory environment in south africa, benefits of south africa for clinical research, selecting a country for your research study.

In our experience, South Africa is presently an underrepresented research country and one that is positioned for growth and inclusion in future clinical trials.
South Africa used to be a commonly utilized country for clinical research, but it became inundated by the number of clinical trial applications which caused bottlenecks in the study approval process. Many investigators and sponsors found the regulatory environment in South Africa to be problematic and inefficient. The old regulatory structure meant that it often took as long as six months to get a study approved. Everything had to pass through the country’s Medicines Control Council (MCC). This entity met infrequently and the session time for drug study consideration was limited. Further, if the MCC required any changes or requested more information, this request would be sent back to the researchers. They then had to respond to the additional requests and wait until the next Clinical Trial Committee session for the response to be reviewed, Final approval could then be granted, or in some instances, more queries were issued. It was not a linear route. The system could not handle the volume of study applications it received. From 2008 to 2018, the timeline for approvals became increasingly slower. In some cases, it took longer than six months to get a green light from the government for a study to proceed. However, that time has since passed. The South Africa regulatory environment has gone through significant changes in recent years. The government there is now much more favorable to clinical research and the country itself offers many advantages.
One of the biggest challenges faced by the South African government was that they needed to change the fundamental ways in which they did things. Documentation is a good example. Throughout this period (of historical inefficiency 2008-2018), the government required two hard copies of the complete submission including site documents. This meant over 1,000 pages needed to be submitted – and that’s not all. They also required 20 copies of the clinical trial application form (CTF-1). This is normally 150 pages. Then, there was the issue of the way study applications were approved. Review and approval could only happen after a full review by the committee and they met only every two months. This fact caused some very limiting issues. For one, the initial submission had to be submitted at least six weeks before the review date so that the materials could be received and processed by the members of the committee. Then, there were the requirements of that committee. In most cases – around 95% – researchers would receive comments from the committee to provide feedback to the CTC (Clinical Trial Committee). These questions would necessitate another meeting of the CTC before receiving approval – which meant waiting another two months until the next committee meeting.
Biotechs and pharmaceutical companies, as an industry, began to demand change and actively lobbied the South African government. Their efforts were enough to call for the involvement of the country’s Minister of Health. South Africa revamped its entire regulatory environment and established SAHPRA to govern the clinical trial review and approval process as well as drug approvals. Limited hard copies are now required – mainly digital . This change meant that provisional approval within four weeks and final approval after 6 weeks following the CTC meeting is now possible. SAHPRA is now consistently providing approvals within 2-3 months or less as mentioned above . This should make South Africa one of the fastest countries, and a promising country candidate for clinical trial consideration.
At Allucent, we have found many reasons to look toward South Africa as a clinical trial site. The speed of the application process is impressive, but that is only a small part of what makes South Africa a clinical research destination.
Cost & Diversity
We have also found that the exchange rate is preferable, allowing sponsor dollars to go further. Patient diversity and disease diversity support several types of research as well. Then, there is the structure of available treatment.
Structure of Treatment
In South Africa, most people receive treatment at state institutions. Less than 30% of South Africans have private medical care. This centralization supplies a patient funnel that is extremely useful for recruitment and study management. It benefits patients too. Clinical trials offer patients more attention and care for their disease than the current state medical structure would offer.
Availability of Strong Sites & Technology
At the same time, South Africa is home to several large oncology centers and SMOs. This streamlines the availability of testing and monitoring equipment. Plus, the medical schools in South Africa are prestigious, large, technologically advanced so finding local investigators who are skilled in the right technologies to successfully conduct and monitor disease progression is also much easier.
Support of Government
Finally, South Africa stands out because it has the support of its government. The country is making efforts to improve population health and putting an emphasis on combating particular diseases. Oncology is a primary area of interest. Tuberculosis and HIV are also major concerns as well as diabetes, cardiac or coronary diseases, and respiratory conditions. Allergen and pulmonary societies are large and mature.
Site selection is a critical part of conducting a successful research study. Different countries have different approval processes and timelines. Understanding these differences can save weeks if not months. Furthermore, patient and disease diversity also come into play. Some places are simply not as diverse as others. As a truly global CRO, Allucent recognizes this and uses these realities to advantage, helping sponsors find the right mix of patients and helping patients access treatment to which they may otherwise not find available. Technological capabilities figure in as well. A good study needs local people with the training to leverage certain technologies and the equipment available to make study testing easy. In our experience, South Africa is a country that is worth considering for many studies.

- Support Forums
- Biostatistics
- CRC Calendar
- Advice, training and tools
- Self-Learning
- What does the CRC do for MMeds?
- Qualitative Methods
- Data Managers' Forum
- Pharmacy Forum
- Project Management Forum
- Regulatory Forum
- Groote Schuur Hospital
- Service Evaluation & Feedback
- Good Clinical Practice
- GCP training service providers
- No-show penalty policy
- Study design and protocol development
- Sponsorship
- Helpful links & docs
- National Registration (NHREC+SANCTR)
- SAHPRA / MCC
- Project and quality management
- Why Data Management?
- Investigational products
- UCT accounts

Staff & Services

A Comprehensive and Practical Guide to Clinical Trials

CRC Video Tour

Services & Facilities
The CRC is an early phase clinical trial unit equipped to conduct first-in-human studies for investigation medicinal products or investigation medical devices. The CRC has its own Research Pharmacy, as well as a Sample Processing Laboratory.

A team of experienced staff can guide and advise researchers in key aspects of conducting their clinical research projects, bringing in other experts where necessary. The CRC helps identify qualified and experienced research team members and provides certain chargeable services and facilities.


Clinical Trials Community Africa Network (CTCAN)
Enabling increased, sustainable, and coordinated clinical trials in africa in africa . broad science area: science innovation pathways -->, science innovation pathways | clinical research & trials community (crtc) | clinical trials community africa network (ctcan).
- Programme Design
The Clinical Trials Community Africa Network (CTCAN) is a network for clinical research stakeholders in Africa that seeks to enable increased, sustainable, and coordinated clinical trials on the continent. The network will build on the progress of the Clinical Trials Community (CTC) platform, which was created to increase the visibility of African clinical trialists and sites while contributing increase in clinical trial investments in Africa.
Many potential clinical sites in Africa need to develop additional capacity before they can take part in Stringent Regulatory Authority (SRA) - quality clinical trials. By entering the Clinical Trials Community Africa Network (CTCAN), sites and laboratories will be able to access a clinical trial preparedness framework. Through the proposed capability development activities, sites and laboratories will be strengthened to generate SRA-quality data, which in turn will be informative for new policies and practices, and lead to earlier access to new treatments.
While governmental and non-governmental organisations (NGOs), pharmaceutical companies, and other public-private sector actors have established important capacity development programmes in Africa, CTCAN draws the different programmes and data together in, or linked to, a single platform. Alignment between these Africa-led programmes and clinical research programmes of clinical trials sponsors will ultimately create a greater, sustainable, and cost-effective impact.
The CTCAN project will lay the foundation for the creation of a coordinated and sustainable umbrella network of clinical trial sites and laboratories capable of undertaking quality clinical research in Africa. The objective is to make the network easily accessible to African and international trial sponsors. A clinical trial preparedness framework will provide the necessary tools to assess site and laboratory capabilities and any potential quality and operational gaps they may have, along with the instruments to mitigate and overcome these gaps.
The CTCAN, an EDCTP-3 supported action, enables an environment that brings more clinical trials to the continent, including large scale clinical trials and disease outbreak response activities. The network will foster coordination in clinical research by providing a platform for all African stakeholders including clinical researchers, regulators, industry partners, and other relevant stakeholders, to define priority diseases for clinical research. The network will also raise awareness of the existing capacity of sites and laboratories across Africa, contribute to the operationalisation and harmonisation of regulatory processes across the region, and strengthen less experienced sites and labs through a clinical trial preparedness framework, while encouraging inter- and cross-continental knowledge and expertise sharing.
Specifically, the CTCAN is designed to:
- Develop an umbrella sub-Saharan African (SSA) network consolidating relevant subnetworks of clinical trial sites and laboratories.
- Supplement the network with epidemiological data associated with the sites and laboratories, clear and operationalised regulatory information required for clinical trial start-up and conduct.
- Develop a framework to enhance capacity and increase clinical trial preparedness.
- Make all this critical information available through a single electronic platform: the Clinical Trials Community platform.
The SFA Foundation is implementing the CTCAN in collaboration with nuvoteQ.io, BIO Ventures for Global Health (BVGH), Global Public Health R&D, a Division of Janssen Pharmaceutica NV, Fondation Merieux, and Medicines Development for Global Health Ltd (MDGH).
By driving and coordinating the scientific aspect of this project, SFA Foundation will underpin, and support comprehensive medical and healthcare strategies developed by African health leaders with the ultimate objective of bringing new innovations to patients faster and enabling African scientists and physicians to discover and develop African solutions for African challenges.
Institutional Leadership
The Programme seeks to create awareness, engagement, and continuity of support for strengthening research management functions in institutions, across generations of leadership and senior academic staff. This will build institutional memory in order to strengthen research management across successive generations of leadership. .
Sustainability
The Programme defines sustainability as the process of change in which the exploitation of resources, prioritisation of investments and technological development are synergistic and enhance current and future human needs and aspirations.
SFA Foundation

The CTCAN partners at their inaugural meeting in Paris, France in November 2023. CTCAN supported by Global Health EDCTP3, enables an environment that brings more clinical trials to Africa. PHOTO| SFA Foundation
How will science policy engagement drive impact:
- If R&I is to deliver maximum impact and positively change the lives of African people and societies, findings from Africa led R&I research programmes should be translated into recommendations that can be implemented within policy and practice in Africa.
- Contribute to efficiency and effectiveness within programmes through best practice.
- Ensure the right questions are formulated, asked, and answered through an African lens.
- Highlight gaps and key points of evidence within context and in an accurate manner to facilitate comprehension and use.
- Promote shared understanding, trust and collaboration for meaningful work that drives change regionally and globally
- Place African experts and policymakers at the centre of science-led policymaking processes in Africa
- Place contextually relevant data and perspectives at the centre of science-led decision-making process in Africa.
Activities being implemented by SPEAR
- Convening African stakeholders to examine and comprehend the policy gaps in AI and Data Science in global health from an African perspective, focusing on but not limited to genomics, clinical trials/drug development, and epidemics/pandemics.
- In collaboration with African partners, identifying the research and development goals of AI and data science for the betterment of global health from an African perspective.
- Engaging the public on AI and Data Science policy
DELTAS Africa Strategic Areas -->
- Enhanced scientific quality: DELTAS Africa produces world-class scientific research that addresses African health and research priorities through scientific discourse and collaborative supervision by promoting collaborations with well-resourced universities, research institutions and think-tanks to strengthen capacity
- Strengthened research leadership capacities: To strengthen scientific research training and build career pathways for scientific researchers, DELTAS Africa focuses on the tertiary and postgraduate training of science students and professionals along a defined career pathway. Training offered by DELTAS Africa programmes is designed to provide individuals at all career stages with the academic support and research facilities they need to develop into world-class researchers
- Strengthened research systems: To cultivate professional environments to manage and support scientific research. This recognises that developing and supporting research requires that researchers have access to skilled administrative support and adequate resources to compete at a global level; and that creating supportive, sustainable environments is crucial to developing research capacity
- Enhanced scientific citizenship / societal engagement: Foster mentorship, leadership and equitable collaboration in science, and engagement with public and policy stakeholders. DELTAS Africa recognises that for research to achieve real impact it needs to be communicated to policymakers and the public. Communicating research findings to policymakers will ensure that the findings inform policy. At the same time, public engagement is also key to raise public awareness and interest in science, increase the uptake of new health policies and treatments, and strengthen relationships with local communities.
" If scientific results are not shared broadly, then the societal benefits are significantly limited. We have a responsibility to ensure that policymakers have access to the best, relevant and up-to-date knowledge available. To achieve Africa’s sustainable development goals, it is critical that policy decisions are informed by contextually relevant evidence”. - Uzma Alam, SPEAR programme lead.
You are using an outdated browser. Please upgrade your browser to improve your experience.

- Independent Research Partners: Who we are
- Independent Research Partners: How we work
- The benefits

Together for the future of health

Welcome to Synexus, your dedicated Clinical Research Centre. We conduct clinical research studies with the help of volunteers like you. By registering with us, we hope that we can find a suitable study for you now, or in the future.
Our Studies by Therapy Area

Vaccine Studies
Immunology & Vaccination
Bones & Joints
Heart & Circulation
Respiratory
Skin Conditions
Digestion & Bowel
Chronic Pain
Mind, Mood & Memory
Kidney & Bladder
Your feedback is vital to the success of our research. Please fill in the form fields below.
Privacy Preference Center
These cookies are necessary for the website to function and cannot be switched off in our systems. They are usually only set in response to actions made by you which amount to a request for services, such as setting your privacy preferences, logging in or filling in forms. You can set your browser to block or alert you about these cookies, but some parts of the site will not then work. These cookies do not store any personally identifiable information.
These cookies allow us to count visits and traffic sources so we can measure and improve the performance of our site. They help us to know which pages are the most and least popular and see how visitors move around the site. All information these cookies collect is aggregated and therefore anonymous. If you do not allow these cookies we will not know when you have visited our site, and will not be able to monitor its performance.
These cookies may be set through our site by our advertising partners. They may be used by those companies to build a profile of your interests and show you relevant adverts on other sites. They do not store directly personal information, but are based on uniquely identifying your browser and internet device. If you do not allow these cookies, you will experience less targeted advertising.
These cookies enable the website to provide enhanced functionality and personalisation. They may be set by us or by third party providers whose services we have added to our pages. If you do not allow these cookies then some or all of these services may not function properly.
Country selection
Regulatory Authority
Scope of assessment, regulatory fees, ethics committee, scope of review, ethics committee fees, oversight of ethics committees.
Clinical Trial Lifecycle
Submission Process
Submission content, timeline of review, initiation, agreements & registration, safety reporting, progress reporting.
Sponsorship
Definition of Sponsor
Site/investigator selection, insurance & compensation, risk & quality management, data & records management, personal data protection.
Informed Consent
Documentation Requirements
Required elements, participant rights, emergencies, vulnerable populations, children/minors, pregnant women, fetuses & neonates, mentally impaired.
Investigational Products
Definition of Investigational Product
Manufacturing & import, quality requirements, product management, definition of specimen, specimen import & export, consent for specimen, requirements, additional resources.
Clinical Trials Registries
- ClinicalTrials.gov listing of studies in South Africa
- International Clinical Trials Registry Platform (ICTRP) consolidated listing of studies in South Africa
Ethics Committees
- Database of institutional review boards/ethics committees registered with the United States Department of Health and Human Services (HHS) Office for Human Research Protections (OHRP)
Funding & Institutions
- World RePORT database of funding organizations, research organizations, and research programs in South Africa
- HHS OHRP database of institutions with approved Federalwide Assurances (FWAs) for the protection of human subjects
South Africa Profile Updated
South africa profile updated in clinregs, south africa: sahpra revises gcp training requirements and issues clinical trial application guidance for public health emergencies, south africa: sahpra issues guidance for clinical trials impacted by covid-19.
Other Regulatory Databases
- United States Department of Health and Human Services (HHS) Office for Human Research Protections (OHRP) International Compilation of Human Research Standards for South Africa
- Health Research Web - South Africa
South African Health Products Regulatory Authority
As stated in the MRSA and ZAF-9 , the South African Health Products Regulatory Authority (SAHPRA) is the regulatory authority overseeing medicines and clinical research, as well as medical devices and radiation safety. A s stated in the MRSA and GRMRSA , SAHPRA is responsible for clinical trial oversight, approval, and inspections in South Africa. The agency grants permission for clinical trials to be conducted in South Africa in accordance with the provisions of the GRMRSA .
Per the MRSA and ZAF-39 , the SAHPRA is an independent, state-owned entity established to oversee the regulation of medicines in South Africa. According to ZAF-39 , this agency is responsible for:
- The regulation of health products intended for human and animal use
- The licensing of manufacturers, wholesalers, and distributors of medicines and medical devices; radiation emitting devices; and radioactive nuclides
- The conduct of clinical trials in a manner that is compatible with national medicines policy
Per the MRSA , SAHPRA is a state-owned entity within the public administration but outside the public service. It acts through a Board appointed by South Africa’s Minister of the National Department of Health (NDOH) . For details on the Board appointments, see ZAF-39 and ZAF-38 .
As described in ZAF-39 and the SA-GCPs , SAHPRA is tasked with regulating (monitoring, evaluating, investigating, inspecting, and registering) all health products. This includes clinical trials, complementary medicines, medical devices, and in vitro diagnostics (IVDs). Its mission is to promote access to health products and protect human and animal health in South Africa through science-based regulatory decisions. Per ZAF-36 , SAHPRA’s Clinical Trial Committee (CTC), within the Clinical Trial Unit, reviews clinical trial applications and bioequivalence studies for human participants and recommends approval of the conduct of clinical trials. SAHPRA also authorizes the importation of unregistered medicine for the purpose of conducting clinical trials. The SA-GCPs also states that SAHPRA is responsible for the following: ensuring efficient, effective, and ethical evaluation or assessment of health products that meet defined standards of quality, safety, efficacy, and performance; ensuring that the process of evaluating or assessing and registering health products is transparent, fair, objective, and concluded in a timely fashion; ensuring periodic re-evaluation and monitoring of health products; and conducting announced and unannounced inspections.
Please note: South Africa is party to the Nagoya Protocol on Access and Benefit-sharing ( ZAF-8 ), which may have implications for studies of investigational products developed using certain non-human genetic resources (e.g., plants, animals, and microbes). For more information, see ZAF-34 .
Contact Information
Per ZAF-35 , SAHPRA’s postal address is:
South African Health Products Regulatory Authority Private Bag X828 Pretoria 0001 South Africa
SAHPRA’s physical address is:
Building A Loftus Park 402 Kirkness Street Arcadia, Pretoria South Africa
As provided in the G-CTA-Electronic and ZAF-36 , the following are the SAHPRA Clinical Trial Unit emails:
New clinical trials application alert, responses to new clinical trial applications and related queries: [email protected] Protocol amendments, responses to amendments and related queries: [email protected] Additional investigators and sites, responses to additional and related queries: [email protected] Bioequivalence (BE) studies, BE amendments, responses to BE studies and related queries: [email protected] Notifications and related queries: [email protected] Individual patient serious adverse events and related queries: [email protected] Guidelines, forms, and related queries: [email protected]
See ZAF-47 for clinical evaluation and management contacts.
In accordance with the GRMRSA , the South African Health Products Regulatory Authority (SAHPRA) is responsible for reviewing and approving all clinical trial applications for an unregistered medicine, and for any new indication or dosage regimen of a registered medicine. The scope of the SAHPRA’s assessment includes all clinical trials (Phases I-IV) and bioequivalence/bioavailability studies. Per ZAF-23 , the review and approval of clinical trial applications by SAHPRA and an accredited ethics committee (EC) may be conducted in parallel.
ZAF-36 states that the SAHPRA’s Clinical Trial Unit (CTU) provides the legal framework for the review of clinical trials and bioequivalence studies for human participants and recommends approval of the conduct of clinical trials. The unit also authorizes the importation of unregistered medicines for the purpose of conducting clinical trials. As per G-GenInfo , the CTU is responsible for the evaluation of clinical trial applications, clinical trial amendments, and adverse event reports arising from a clinical trial.
Clinical Trial Review Process
Per ZAF-36 , the CTU of SAHPRA receives, processes, and evaluates clinical trial applications and any subsequent amendments for approval to conduct a study within South Africa. Researchers must submit a completed application and the prescribed fee on predetermined dates ( ZAF-11 ). The proof of delivery, proof of payments, and cover page must be sent to SAHPRA via email.
As stated in ZAF-36 , the CTU completes a preliminary screening of the application and sends an official letter to the applicant with the outcome and follow-up questions on a screening checklist. As indicated in ZAF-23 , incomplete documentation or sub-standard submissions will be rejected. Additionally, applications submitted without clinical trial insurance will be rejected. Applicants will be allowed a maximum of two (2) rounds of queries to respond to, and if the responses are not satisfactory the application will be rejected. Per ZAF-36 , if an application is rejected, no response is required; the screening checklist should be used as guidance for resubmission during the next review cycle. Next, the CTU’s Clinical Trial Committee (CTC) (which includes an expert committee of specialists, as needed) reviews the proposed clinical trials pursuant to the schedule on SAHPRA’s website. (See ZAF-11 for 2024 dates). Clinical trial reviews will result in one (1) of the following outcomes:
- Category 1A: Approved; no items pending
- Category 1B: Approved; ethics approval pending
- Category 2A: Not approved; for approval by in-house evaluators, 1-2 or more items outstanding as deemed by the committee
- Category 2B: Not approved; for approval by the original evaluator and in-house if a need arises
- Category 3: Not approved; items outstanding to be discussed at the next CTC meeting
- Category 4: Not approved; for referral for specialist opinion
- Category 5: Not approved – technical/scientific deficiencies; applicant to resubmit for the next cycle
- Category 6: Rejected due to administrative and technical items outstanding; applicant to resubmit for the next cycle
If an applicant would like to request a meeting with the CTC, the request should be submitted through the SAHPRA Chief Executive Office pursuant to the procedures in the G-ConsultMtg .
Other Considerations
Per the G-Capacity , SAHPRA will also review clinical trial applications for evidence of plans to build capacity at each study site as well as enhancing research activities and skills of professionals from historically disadvantaged groups. See G-Capacity for detailed information on actions that will comply with this requirement.
In addition, see G-Clin for South Africa's use of a “reliance model” to register medicines based on clinical trial data from other regulatory authorities.
Per the MRSA , the South African Health Products Regulatory Authority (SAHPRA) is authorized to make regulations to collect fees for its various medicine regulatory functions. As delineated in the MRSA-Fees and ZAF-37 , applicants are responsible for paying several non-refundable fees to submit a clinical trial application. MRSA-Fees delineates the following fees:
For a clinical trial application for the authorization of the use of unregistered medicines:
- Clinical trial application (safety and efficacy): South African Rand (R)32 400
- Clinical trial application (bioequivalence study): R30 400
- Clinical trial application (postgraduate study): R10 800
- Any other clinical trial application: R5 000
For amendments to clinical trials:
- Technical amendment applications: R7 000
- Administrative amendment applications: R4 100
- Any other application except for the purpose of performing a clinical trial: R350
For licenses:
- New manufacturing license: R25 200
- New import/export license to the holder of certificate of registration: R15 000
- Renewal of manufacturing license: R22 000
- Renewal of import license to the holder of the certificate of registration: R9 200
- Renewal of export license to the holder of the certificate of registration: R9 200
- Annual retention of all licenses: R4 200
For inspections to assess the quality, safety, and efficacy of medicines:
- Local and international manufacturing sites: R1 600 per hour
- Local and international clinical trial sites: R1 600 per hour
Payment Instructions
Per the G-SAHPRAFees , when making payments, applicants should follow these guidelines:
- Applicants should submit a cover page that identifies the services requested using the template provided in ZAF-37
- Payments should be referenced in accordance with the SAHPRA Fee Categorization Guideline (Annexure A of G-SAHPRAFees )
- If the applicable bank limits reference spacing, follow the sequence listed in Annexure A as far as the limitation allows; spacing and dashes (/) may be omitted
- Fee payments may be transferred directly into the bank account of SAHPRA via an electronic or manual deposit process
- No check payments will be accepted
- For administrative control purposes, applicants should make one (1) payment per service
- Payment should only be made once the application and required dossiers are ready for submission
- Payments do not have to be made upon request of an application number; however, the applications and required dossiers should be submitted within a reasonable time upon receipt of an application number or as specified in the relevant application guidelines
- As soon as the fee payment has been made, the proof of payment and cover page should be attached and sent via email to SAHPRA Finance at [email protected] , and the relevant unit(s) processing the application should be copied on the email.
- If the proof of payment has not been submitted, or no details to identify the payment reference as per the G-SAHPRAFees have been provided, and any further attempts to clear these payments fail after 12 months, any liability for SAHPRA to refund these payments will be forfeited
- If a payment has been received without an application, the applicant will be notified to submit the required application within 14 working days, failing which, the amount will be forfeited
- Requests for refunds should be submitted in line with Annex B in the G-SAHPRAFees
- Payment and pro forma invoice queries and requests can be directed to [email protected] or 012 501 0323
- See the G-SAHPRAFees for details on special requests for extensions to the deadline
Per the G-SAHPRAFees , the bank and account details are as follows:
Account name: South African Health Products Regulatory Authority Special Name: The Medicines Control Council Account type: Cheque/Current Account Account number: 40-5939-2080 Bank: ABSA Bank Branch Code: 632005 Bank physical address: 240 Vermeulen Street, Pretoria, 0001, South Africa Swift Code: ABSAZAJJ
Fee payment questions can be directed to [email protected] or 012 501 0323.
Per ZAF-51 , ethics committees (ECs) in South Africa are governed by the National Health Research Ethics Council (NHREC) , which is a statutory body established under the NHA . According to ZAF-52 , NHREC gives direction on ethical issues relating to health and develops guidelines for the conduct of research involving humans and animals. As delineated in the NHA , the G-EthicsHR-ZAF , and the SA-GCPs , all ECs are required to register with the NHREC in order to undertake the ethical review of a clinical study.
The NHA requires that every institution, health agency, and health establishment at which research is conducted establish an EC or have access to an independent EC. The EC must be registered with the NHREC. The SA-GCPs note that the NHREC accredits and audits the ECs.
Ethics Committee Composition
As delineated in the SA-GCPs and the G-EthicsHR-ZAF , an EC must consist of members who collectively encompass the qualifications and experience required to review and evaluate the scientific, medical, and ethical aspects of all proposed research studies.
The G-EthicsHR-ZAF indicates that an EC should comprise:
- Members who have documented proof of research ethics training, refreshed at least once within the period of appointment
- At least nine (9) members
- At least one (1) layperson
- At least one (1) member with knowledge of, and current experience in the professional care, counselling, or health-related treatment of people; such a medical practitioner, psychologist, social worker, or nurse
- At least one (1) member with professional training and experience in qualitative research methodologies
- Members with professional training and experience in quantitative research methodologies
- A member with expertise in bio-statistics
- A member with expertise in research ethics
- At least one (1) member who is legally qualified
Terms of Reference, Review Procedures, and Meeting Schedule
Per the G-EthicsHR-ZAF , an institution or organization must select EC members according to prescribed recruitment and appointment procedures. Members must receive a formal notice of appointment and assurance that they will be legally protected with respect to any liabilities that may arise during their term. EC quorum should be a simple majority, and where the number of members is more than 15, the quorum may be 33%. An EC must also establish and record written procedures to address several administrative issues including meetings, agenda/minutes preparation, research protocol presentations, application registration, protocol submission requirements, review and decision notification process, adverse event reporting, protocol amendment reporting, and end-of-trials review. A reasonable term of office is between two (2) and four (4) years, renewable twice, after which the person should stand down for at least one (1) term. Further, EC members and researchers are expected to familiarize themselves with the institutional documentation as well as national and international research ethics guidelines and should have documented proof of such familiarity. Training of all EC members is critical, especially for ECs that review high-risk research. Training and refresher courses should be available, and EC members should produce, at least once during a term of appointment, evidence of recent training. This ensures that both expertise and responsibility are distributed and encouraged in a range of members, and that institutional memory is accumulated. The SA-GCPs stipulate that EC members who review clinical trial proposals should have research ethics training and good clinical practice training, evidenced by certificates issued in the last three (3) years.
Per the SA-GCPs , the EC should retain all relevant records for a period of at least three (3) years or as per institutional requirement, whichever period is longer, after completion of the trial and make them available upon request from the applicable regulatory authority. The G-EthicsHR-ZAF indicates that ECs should keep written records of all research protocols received for review in the form in which they were approved. Electronic records are acceptable if the signatures are properly documented and included in the record. EC records must provide a reliable and authoritative record of the EC’s business that will stand up to scrutiny in the event of queries, conflicts, and audits.
Per the SA-GCPs , clinical trials should be conducted in accordance with all ethical principles outlined in the Declaration of Helsinki ( ZAF-44 ) and consistent with good clinical practice and other applicable regulatory requirements. In accordance with the NHA , the SA-GCPs , and the G-EthicsHR-ZAF , ethics committees (ECs) must evaluate the ethical and scientific rigor of all research studies to be conducted in the country. An EC’s primary responsibilities are to (Note: the regulations provide overlapping and unique elements so each of the items listed below will not necessarily be in each source):
- Review protocols to ensure that research involving human participants has scientific merit and will promote health, and prevent or cure disability and disease; in addition, ensure the research has social merit in light of South Africa’s research priorities or is otherwise justified
- Ensure clinical trials are governed by the ethical principles of beneficence and non-maleficence, distributive justice (equity), and respect for persons (dignity and autonomy)
- Grant approval for research where the protocols meet the ethical standards of the institution, agency, or establishment
- Determine whether and why randomization is relevant, and how this is addressed
- Evaluate the appropriateness of the inclusion/exclusion criteria and the recruitment process in the South African context
- Ensure the feasibility of obtaining meaningful results with the lowest possible risk of harm for participants and whether the risk of harm is appropriately weighed against anticipated benefits for participants or the class of persons from which they are drawn; high risk of harm may be justifiable where the anticipated benefit is of high importance to increase relevant knowledge and appropriate mitigating measures are in place to minimize harm to participants; and attention must be given to harms and benefits beyond the life of the trial itself, especially in respect to early phase studies and (pharmacovigilance) surveillance for chronic and life-threatening conditions
An EC must also pay special attention to protecting the welfare of certain classes of participants deemed to be vulnerable (See the Informed Consent topic for additional information about these populations).
Role in Clinical Trial Approval Process
Per the G-EthicsHR-ZAF , the SA-GCPs , and the NHAParticipants , the principal investigator (PI) or the sponsor must submit a clinical trial application to both the South African Health Products Regulatory Authority (SAHPRA) and a registered EC for review and approval before a study may commence. Per ZAF-23 , the review and approval of clinical trial applications by SAHPRA and an accredited EC may be conducted in parallel.
The G-EthicsHR-ZAF indicates that after the deliberative review process, the EC should approve, require amendment to, or reject a research protocol. In considering a research protocol, the EC may seek assistance from experts. EC decisions should be recorded in writing. A decision to approve should include the conditions (e.g., the duration of the approval, the reporting requirements, etc.). Reasons for a decision to require an amendment or to reject a research protocol should be recorded. Outright rejection should be avoided if a researcher can be advised to improve the protocol. Researchers should be encouraged to address the concerns and improve their protocols. ECs should require researchers to report immediately if a project is terminated or suspended before the anticipated date of completion. ECs should require researchers to report immediately anything that might warrant reconsideration of ethical approval of the protocol, including but not limited to:
- Serious or unexpected adverse effects on participants
- Proposed changes in the protocol
- Unforeseen events that might affect continued ethical acceptability of the project
Per the G-EthicsHR-ZAF , ECs may, at their own discretion, recognize prior review and approval of a research protocol by another registered EC to avoid duplication of effort. Reciprocal recognition means that two (2) or more registered ECs decide to recognize each other’s prior review. ECs that recognize prior review in this manner must determine the nature of the documents to be filed locally, which must, at minimum, include a copy of the approval letter from the other ECs. In addition, ECs may establish procedures for expedited review for research that poses no more than minimal risk of harm to participants.
The SA-GCPs requires the EC’s approval of the following before the clinical trial may begin: protocol and any amendments; case report form, if applicable; informed consent form(s); any other written information to be provided to the participants; advertisement for participant recruitment (if used); participant compensation; and any other documents given approval/favorable opinion.
The SA-GCPs mandate that the sponsor receive confirmation of EC review from the investigator(s) or institution(s). The sponsor must receive the following information prior to the trial’s commencement:
- The name and address of the relevant EC registered with National Health Research Ethics Council (NHREC) , with its documented approval
- If EC approval is conditional on required modifications, a copy of the modification(s) made and the date the final approval was granted by the EC
- Documentation and dates of any EC re-approvals/re-evaluations
As delineated in the G-EthicsHR-ZAF , ECs have the right to monitor the research it approves, and researchers should provide appropriate information to the EC to facilitate monitoring, including alerts and investigator brochures. The frequency and type of monitoring should reflect the degree and extent of risk of harm to participants or animals. ECs may recommend and adopt any additional appropriate mechanism for monitoring.
Per ZAF-20 , if there is an amendment to the protocol, the sponsor must notify the EC and get its approval. This approval should be sent to the SAHPRA using the Application for Protocol Amendment to an Approved Trial ( ZAF-20 ).
Based on the G-EthicsHR-ZAF , ethics committees (ECs) may independently decide whether to charge fees for a protocol review. The G-EthicsHR-ZAF states that an EC should establish and record working procedures concerning fees charged, if any. Researchers without affiliation to an institution or organization with an EC should approach a registered EC to request it to review their health research protocols. If the EC is willing to review external applications, a fee for service may be levied.
Per ZAF-51 , ethics committees (ECs) in South Africa are governed by the National Health Research Ethics Council (NHREC) , which is a statutory body established under the NHA . As delineated in the NHA , the NHREC was created by the Minister of Health to provide ethical oversight of clinical research and to safeguard the rights and welfare of human participants involved in clinical studies. According to ZAF-52 , NHREC gives direction on ethical issues relating to health and develops guidelines for the conduct of research involving humans and animals. Further, NHREC upholds the principle that research involving human participants is based on a moral commitment to advancing human welfare, knowledge, and understanding, and to exploring cultural dynamics, especially in large-scale trials conducted in developing countries. Of fundamental importance is the duty to conduct scientifically sound research while acting in the participant’s best interests and respecting and protecting the participant’s autonomy.
As delineated in the NHA , the SA-GCPs , and the G-EthicsHR-ZAF , the NHREC’s core responsibilities center on promoting, ensuring, and monitoring compliance by ECs. According to ZAF-52 , the functions of the NHREC include:
- Determine guidelines for the functioning of ECs
- Register and audit ECs
- Set norms and standards for conducting research on humans and animals including clinical trials
- Adjudicate complaints about the functioning of ECs
- Refer to the relevant statutory health professional council matters involving the violation or potential violation of an ethical or professional rule by a health care provider
- Institute such disciplinary action as prescribed
- Advise the national department and provincial departments on any ethical issues concerning research
Registration, Auditing, and Accreditation
As delineated in the NHA , the G-EthicsHR-ZAF , and the SA-GCPs , all ECs are required to register with the NHREC in order to undertake the ethical review of a clinical study. The application to register an EC is available at ZAF-53 . ZAF-54 states that the EC registration is recorded and publicly listed by the NHREC. The annual report form that ECs must submit to NHREC is available at ZAF-54 . Per the SA-GCPs , the NHREC accredits and audits the ECs.
As delineated in the SA-GCPs , the sponsor and the investigator must obtain approval from the South African Health Products Regulatory Authority (SAHPRA) and a registered ethics committee (EC) to begin a clinical trial in South Africa. Per ZAF-23 , the review and approval of clinical trial applications by SAHPRA and an accredited EC may be conducted in parallel. Per ZAF-20 , the same process applies to the review and approval of an amendment to the protocol.
Regulatory Submission
Per ZAF-36 , researchers must submit a completed application ( ZAF-23 ) and the prescribed fee on predetermined dates ( ZAF-11 ) and obtain proof of delivery. The proof of delivery, proof of payments, and cover page must be sent to SAHPRA via email. The G-CTA-Electronic delineates the electronic submission and communication process in SAHPRA’s Clinical Trial Unit (CTU). For new clinical trial applications (excluding bioequivalence studies), upon submission at SAHPRA Reception, applicants are requested to alert the CTU via e-mail at [email protected] and include a copy of the proof of delivery, proof of payment, and proof of insurance. In the subject of the e-mail, provide type of application, protocol number, SAHPRA predetermined cycle (see ZAF-11 ), and email number in case of multiple emails (e.g., “email 1 of 5”). Note that the submission email must include organized zipped folders for various sections of the clinical trial application. Individual site documents for each staff member must be uploaded into one (1) document and labelled with the staff name and arranged in folders according to the site which they belong to.
Per G-CTA-Electronic , to respond to SAHPRA’s screening checklist or to CTU’s expert committee review, the applicant must submit all responses by e-mail to [email protected] and include labelled attachments to the required documents. In the subject of the email, the applicant should provide the type of application, protocol number, and SAHPRA database tracking number. Responses to the CTU’s expert committee recommendations can be in MSWord or PDF formats. All other accompanying documents should be in PDF format v1.4, 1.5, 1.6, or 1.7 and legible with the Acrobat Reader search plugin or any other freeware viewer. PDF files should be saved as “Optimized” to reduce the size and allow faster opening when viewed online. The use of additional software to navigate and work with the files is not acceptable. If PDF files are not produced from an electronic source document but from scanned paper, readability and file size should be balanced; the following is recommended: resolution 300 dpi (photographs up to 600 dpi), avoid grayscale or color where possible, use only lossless compression techniques. The file must be searchable (OCR scanned). In addition, the maximum size of documents allowed per e-mail is 5 MB. As per arrangement with CTU, in case of a big file of documents and documents that need to be couriered, the waybill should indicate the type of application, protocol number, and SAHPRA database tracking number.
Per G-CTA-Electronic , for bioequivalence studies, the application and accompanying documents should be emailed to [email protected] . The clinical trial application form should be in MS Word format and all other accompanying documents in PDF, as described above. As per arrangement with CTU, in case of a big file of documents and documents need to be couriered, the waybill should indicate the type of application, protocol number and SAHPRA database tracking number. The email subject should include the type of application, protocol number, and SAHPRA database tracking number. See the G-CTA-Electronic for specific examples of labeling the emails.
Per the G-CTAPHEmerg , during a public health emergency, applicants should use the modified clinical trial application form in G-CTAPHEmerg . This form recognizes the constraints on the availability of information posed by the emergency. SAHPRA may accept clinical trial applications with reduced information together with a commitment to update and complete the required information as soon as possible. However, all documents submitted must be organized with zipped folders according to the checklist in G-CTAPHEmerg and correctly labelled to ensure easy validation by SAHPRA (See the Submission Content and Emergencies sections for more details).
The G-CTA-Electronic provides instructions on submitting protocol amendments during the conduct of clinical trials, for additional investigators and sites during the conduct of clinical trials, bioequivalence studies, notifications and notification studies, and individual serious adverse events. The applicant must submit to SAHPRA the application for amendment to an approved trial ( ZAF-20 ), as well as notify and get EC approval. (Also see Site/Investigator Selection and Safety Reporting sections for information about these submittal processes.)
The G-CTA-Electronic and ZAF-23 state that the clinical trial application must be sent to SAHPRA in a submission email (per directions above). However, ZAF-1 provides the following address for delivery of clinical trial applications to SAHPRA Reception:
South African Health Products Regulatory Authority SAHPRA reception – 2nd floor Loftus Park, Building A 402 Kirkness St, Arcadia Pretoria, 0007 South Africa
Per ZAF-1 , upon receipt of the clinical trial application at SAHPRA Reception, an acknowledgement of receipt in the form of a stamp and signature will be issued. The waybill from a courier company does not suffice as proof of delivery. SAHPRA’s CTU requires a document, referred to as the ‘stamp page,’ which includes the SAHPRA trial reference number, protocol number, and study title. This document will then be date-stamped and signed by SAHPRA’s Administrative Department and returned as proof.
As per the GRMRSA , all applications and supporting data submitted to the SAHPRA should be presented in English. Original documents that are not in English must be accompanied by an English translation.
Ethics Review Submission
Each EC has its own required submission procedures, which can differ significantly regarding the number of copies to be supplied and application format requirements. Refer to each EC’s website for specific submission procedures (Note: ECs are referred to as health research ethics committees (HRECs) in South Africa).
Regulatory Authority Requirements
As per ZAF-23 , the following documentation must be submitted to the South African Health Products Regulatory Authority (SAHPRA) :
- The clinical trial application form ( ZAF-23 )
- Two (2) cover letters (one (1) signed in PDF and one (1) in MS-Word format)
- Two (2) completed copies of the clinical trial application (one (1) signed in PDF and one (1) in MS-Word format) ( ZAF-23 and ZAF-20 (for amendments))
- Patient information leaflets (PILs) and informed consent forms (ICFs); include standardized SAHPRA contact details (Annex 1 of ZAF-23 )
- Copy(ies) of recruitment advertisement(s) (if applicable) and questionnaires
- Investigator’s Brochure (IB)/SAHPRA and other regulatory authorities’ approved professional information (Package insert(s))
- Summary of previous trials with the investigational product(s) (IP(s)), if applicable
- Certificate of analysis of the product
- Signed investigator(s) Curriculum Vitae(s) (CV) in SAHPRA format (Annex 2 of ZAF-23 )
- Signed declaration(s) by all investigator(s) (Annex 3 of ZAF-23 )
- Signed joint financial declaration by sponsor and principal investigator (PI) or national PI (Annex 4 of ZAF-23 )
- Signed declaration by applicant and national PI
- Signed declaration by national PI (See page 4 and Annex 3 ( ZAF-23 )
- Signed declaration by sub-investigators (Annex 5 of ZAF-23 )
- CV(s) and signed declaration by regional monitor(s) (Annexes 2 and 6 of ZAF-23 )
- Proof of application to register the trial on the South African National Clinical Trials Register (SANCTR) ( ZAF-48 )
- Active insurance certificate for clinical trial
- Proof of sponsor indemnity for investigators and trial site(s) (Annex 7 of ZAF-23 )
- Active Good Clinical Practice (GCP) Certificates
- Workload forms for investigators (Annex 8 of ZAF-23 )
- Proof of registration with professional statutory bodies
- Proof of professional indemnity (malpractice insurance) of trialist(s)
- Ethics committee (EC) approval letter or copy of letter submitted to EC
- Study budget
- Electronic copies of key peer reviewed publications following International Committee of Medical Journal Editors (ICMJE) recommendations to support the application (if applicable)
- Proof of payment (bank validated)
- Certificate of good manufacturing practice (GMP) for manufacture of the IP(s) (including placebo and comparator)
- Evidence of accreditation/certifications of the designated laboratories
- Data Safety Monitoring Board charter and composition (where applicable)
See ZAF-36 for additional information on submissions. For phase IV trials of approved products, the applicant must notify SAHPRA following the instructions provided in ZAF-17 .
ZAF-20 delineates the contents and requirements for submitting an application for protocol amendment to an approved clinical trial.
Per the G-CTAPHEmerg , SAHPRA states that during a public health emergency, new and experimental treatments may become necessary and clinical trials are essential to provide the evidence to develop appropriate policies for patient treatments. Under these circumstances, there may be limited information available. However, applications need to contain a certain minimum of information to enable a meaningful evaluation and regulatory decisions. To address this, SAHPRA provides an information grading system in the G-CTAPHEmerg wherein required information is labelled. Applicants must attempt to provide the information listed below and justify when this is not available. The required information is graded as follows:
- Essential – Application will not be considered without this
- Important – Necessary information that must be provided later and must be justified if not available
- Not essential – May be omitted from this preliminary application
All incomplete information must be explained, justified, and provided to SAHPRA as a complete application ( ZAF-23 ), when available. This means that repeat evaluations of an application may be necessary.
Ethics Committee Requirements
Each EC has its own application form and clearance requirements which can differ significantly regarding the number of copies to be supplied and application format requirements. However, the requirements list provided below is basically consistent across all South African ECs.
The following list was compiled from ZAF-24 , ZAF-22 , ZAF-45 , ZAF-42 , and ZAF-49 , to exemplify the common elements shared by the various application forms:
- Cover letter
- Completed EC-specific application form
- Protocol synopsis
- PIL(s) and ICF(s) and process for obtaining informed consent
- Separate assent form required for adolescents/children under the age of 18 (See Children/Minors section for additional information)
- IB and package insert(s) (if applicable)
- SAHPRA approval letter or letter of application and notification
- Approval letter from institution’s scientific committee (if applicable)
- Copy of completed clinical trial application signed by all participating investigators
- All questionnaires and diaries to be used in the study
- Advertisement(s) (if applicable)
- Trial site information (address, telephone numbers, PI names, etc.)
- Trial payment schedule and budget schedule per site/draft financial contract and additional funding details
- Proof of submission fees payment
- Current investigator(s) CVs
- GCP training certificates for PIs and subinvestigators
- Information on registration with SANCTR ( ZAF-48 )
- Declaration of trialists (PI and sub-investigators) in SAHPRA format
- Insurance certificate
Further, per the MTA-Human , all the providers and recipients of human biological material for use in research or clinical trials under the auspices of ECs must use the “Material Transfer Agreement of Human Biological Materials” in MTA-Human . The agreement must be signed by the research institution’s authorized representative and the EC. (For additional details, see Specimens topic .)
Clinical Protocol
As delineated in ZAF-23 and the SA-GCPs , the clinical protocol should contain the following information (Note: the sources provide overlapping and unique elements so each of the items listed below will not necessarily be in each source.):
- General information
- Background information
- Study rationale and motivation
- Trial objectives, purpose, and endpoints (with justifications)
- Trial design and methodology
- IP information
- Participant eligibility, selection, and withdrawal
- Participant treatment
- Efficacy assessment
- Safety assessment
- Direct access to source data/documents
- Quality control/quality assurance
- Data and safety monitoring plan
- Data handling/recordkeeping
- Statistical measures
- Financing/insurance
- Publication policy
Per the SA-GCPs , the protocol must also provide details on ethical and administrative issues, including how the following matters are addressed:
- Compliance of multi-center/national trials with all South African regulatory requirements
- The trial design must be customized appropriately for the local setting to ensure that local realities are considered and appropriately integrated into the design
- For multi-national trials, whether a reasonable proportion of significant project team members, including scientists and health care professionals, are South African researchers, including those from previously disadvantaged backgrounds
- If South Africa is selected as a clinical trial site but the country of origin or other high-income countries are not, an explanation and reason for this with a clear ethical justification
For detailed information on protocol elements, please refer to ZAF-23 and the SA-GCPs .
Based on ZAF-23 and the SA-GCPs , the review and approval of clinical trial applications by the South African Health Products Regulatory Authority (SAHPRA) and an accredited ethics committee (EC) may be conducted in parallel. The applicant must notify each regulatory body of the other’s approval once it has been received.
Regulatory Authority Approval
In general, per ZAF-36 , SAHPRA’s Clinical Trial Unit (CTU) aims to process new applications and issue a screening checklist within three (3) weeks of receipt. After that, the expert Clinical Trials Committee (CTC) recommendations will be sent within 10 weeks of the submission due date. There are cases where this turnaround time might be prolonged, such as an unfamiliar investigational product which may be referred to external reviewers or other SAHPRA committees for input.
Per ZAF-1 , during the preliminary screening, the CTU screens the application and sends an official letter to the applicant with the outcome and follow-up questions on a screening checklist. The applicant receives the screening checklist within 15 working days after application submission. The applicant must respond within seven (7) working days after receipt of the screening review.
Next, the CTC reviews the proposed clinical trials. ZAF-11 provides the dates of the 2024 CTC meetings and the SAHPRA submission due dates. It is advisable to submit clinical trial applications before these due dates. Once the reviewer approves the application, the CTC presents the committee’s/reviewer’s recommendations to the SAHPRA. ZAF-1 states that applicants receive a response within 10 working days from the CTC meeting, and they must send an answer within seven (7) working days after receipt of comments. If an applicant would like to request a meeting with the CTC, the request should be submitted through the SAHPRA Chief Executive Office pursuant to the procedures in the G-ConsultMtg .
Ethics Committee Approval
As earlier stated, an applicant must also submit the clinical trial application for review and approval by an accredited local EC. Review timelines vary per an individual EC’s procedures.
In addition, as described in the G-EthicsHR-ZAF and ZAF-6 , all clinical trials must obtain site-specific provincial and/or hospital approval to assess the impact the clinical trial will have on the resources of the establishment hosting the trial.
In accordance with the GRMRSA , the SA-GCPs , the G-EthicsHR-ZAF , and the NHAParticipants , a clinical trial can only commence in South Africa once an applicant receives approval from the South African Health Products Regulatory Authority (SAHPRA) and from an accredited local ethics committee (EC). There is no waiting period required following the applicant’s receipt of these approvals.
In addition, the principal investigator (PI) for each study site must be a South African-based scientist (resident of South Africa), and should have the appropriate qualifications, training, and experience to assume responsibility for the proper conduct of a trial. The trial must be conducted in compliance with the SA-GCPs , the G-EthicsHR-ZAF , and the GRMRSA . Also, per the SA-GCPs , all clinical trials must be conducted in a laboratory complying with Good Laboratory Practices (GLP). See ZAF-46 for the World Health Organization (WHO) ’s handbook on GLPs.
Per the SA-GPPs , pharmacists must be involved in clinical trials, including for example, assisting in the development of protocols, overseeing medicine supplies, monitoring administration protocols, and maintaining registries.
Clinical Trial Agreement
According to the SA-GCPs , all parties involved in the conduct of a trial should be familiar with guidance in the International Council for Harmonisation's Guideline for Good Clinical Practice E6(R2) ( ZAF-27 ) and other international guidelines. Before the trial begins, a sponsor must prepare a written agreement. The agreement must be signed by the sponsor and the PI, and any other parties involved (e.g., institutions and contract research organizations) with the trial to confirm the contract terms. Both the sponsor and the PI must commit to providing safety information between each other. The sponsor should also obtain the investigator's agreement to:
- Conduct the trial in compliance with the SA-GCPs , the SAHPRA requirements, ZAF-27 , and the EC approved protocol
- Comply with data recording/reporting procedures
- Permit monitoring, auditing, and inspection
- Retain the trial-related essential documents until the sponsor informs the investigator(s) and institution(s) that these documents are no longer needed
In addition, per the SA-GCPs , the financial aspects of the trial should be documented in the agreement. A declaration must be signed by the sponsor and PI stating that sufficient funds are available to complete the study. The sponsor is also responsible for securing agreements to ensure direct access to all trial-related sites, source data/documents, and reports for the purpose of monitoring and auditing by the sponsor, and inspection by domestic and foreign regulatory authorities.
Clinical Trial Registration
According to the SA-GCPs , NHAParticipants , and ZAF-32 , the PI or the sponsor must enter the trial information in the South African National Clinical Trials Register (SANCTR) ( ZAF-48 ). The SA-GCPs indicates that the National Department of Health (NDOH) then issues a unique SANCTR National Register Number. ZAF-32 has instructions for registering either online or via email.
ZAF-48 states that SANCTR fulfills the requirements of the International Committee of Medical Journal Editors (ICMJE) publication mandates and has a formal partnership with the Pan African Clinical Trials Registry ( ZAF-50 ), which is recognized by the WHO.
Safety Reporting Definitions
In accordance with the SA-GCPs , the G-EthicsHR-ZAF , and the G-SafetyRpt , the following definitions provide a basis for a common understanding of South Africa’s safety reporting requirements:
- Adverse Event/Experience (AE) – Any untoward medical occurrence that may present during treatment with a medicine, but which does not necessarily have a causal relationship with this treatment
- Adverse Drug Reaction or Adverse Reaction (ADR) – A noxious and unintended response to a medicine in humans or animals, including lack of efficacy, and which occurs at any dosage and can also result from overdose, misuse, or abuse of a medicine
- Serious Adverse Event (SAE) or Serious Adverse Drug Reaction (SADR) – Any untoward medical occurrence that at any dose: results in death, is life-threatening, requires patient hospitalization or prolongation of existing hospitalization, results in persistent or significant disability or incapacity, or is a congenital anomaly or birth defect
- Unexpected Adverse Drug Reaction – One in which the nature, specificity, severity, and outcome is inconsistent with the applicable product information (i.e., with the approved package inserts for registered medicines, the investigator’s brochure, or other product information for unregistered medicines being used)
Furthermore, ZAF-30 provides clarification on the definition of a serious suspected unexpected adverse reaction (SUSAR), which is a reporting requirement in the updated G-SafetyRpt . Per ZAF-30 , a SUSAR is an adverse reaction that is unexpected but suspected to be drug related. It must fulfil the criteria for “serious” as per the definition of SAEs. In addition, all SUSARs are SAEs but not all SAEs are SUSARs.
Per the G-EmergencyProc , all clinical trial sites must have an emergency standard operating procedure that should be available for inspection by the South African Health Products Regulatory Authority (SAHPRA) . In addition, each clinical trial site should have adequately trained investigators to manage medical emergencies. Further, there must be an emergency 24-hour contact number for trial participants who experience an unexpected AE.
Safety Reporting Requirements
Investigator Responsibilities
As specified in the SA-GCPs and the G-EthicsHR-ZAF , the principal investigator (PI) must inform the sponsor immediately, or within the time specified in the protocol, of any serious and/or unexpected AEs occurring during the study. The initial reporting form and any relevant follow-up information should be sent to the sponsor. The G-SafetyRpt directs the investigator to report AEs to the sponsor in a manner defined in the protocol. Per the SA-GCPs , AEs and/or laboratory abnormalities identified in the protocol as critical to safety evaluations must be reported to the sponsor in accordance with the reporting requirement and within the time periods specified in the protocol. In the case of participant deaths, the PI must supply the sponsor, the ethics committee (EC), and SAHPRA with any additional information, as requested. The initial and follow-up reports must identify the affected participants by the participant identification code.
Sponsor Responsibilities
As delineated in the GRMRSA and the G-EthicsHR-ZAF , the sponsor is required to report all expected or unexpected SAEs/SADRs on an expedited basis to all concerned parties, including the investigator(s) and institution(s), the SAHPRA, and the ECs. Pursuant to the G-SafetyRpt , the sponsor is required to submit the following safety reports to SAHPRA:
- Reports of SUSARs occurring in the clinical trial using the SAHPRA SAE form ( ZAF-19 ), CIOMS form ( ZAF-15 ), or Annex B of G-SafetyRpt
- Reports of all SUSAR and trends occurring with the investigational product (IP) in South Africa
- Six-month progress report
- Annual Development Safety Update Reports (DSURs) that includes information gathered from all clinical experience with the IP, whether in South Africa or elsewhere
- Final Progress Report
- Final Study Report
The SA-GCPs states that the sponsor is responsible for performing an ongoing safety evaluation of the IP and must promptly provide written notification to the investigator and SAHPRA of findings that may adversely affect the safety of participants or the conduct of the trial, and/or change the EC's approval to continue the trial. The commitment to provide safety information must be included in the clinical trial agreement signed between the sponsor and the investigator.
The G-SafetyRpt delineates the following reporting timeframes:
- The sponsor should initially report all fatal or life-threatening SAEs in local reports within seven (7) calendar days after first knowledge, using CIOMS format ( ZAF-15 )/SAHPRA SAE form ( ZAF-19 ). The follow-up report should be submitted within an additional eight (8) calendar days.
- All fatal or life-threatening SAEs in foreign reports should initially be reported within 30 calendar days after first knowledge by the sponsor. The follow-up report should be submitted within an additional six (6) months as part of the progress report. If the SAEs result in premature study closure, the reporting times are shorter—seven (7) days for the initial report and within an additional eight (8) days for the follow-up report. These reports should be in a line listing format. Note that these reporting requirements also cover foreign reports of “special concern,” which is a significant safety issue defined for each clinical trial that requires urgent attention from the regulatory authority. An adverse reaction of special concern from a foreign jurisdiction should be based on the decision of its regulatory authority. A safety issue leading to international regulatory action is considered to be significant at all times and hence reportable.
- Local reports of other serious events (unexpected, not fatal or life threatening) within 15 calendar days of the event and every six (6) months in the CIOMS format ( ZAF-15 )/SAHPRA SAE form ( ZAF-19 )
- A line listing of all local reports—serious (unexpected and expected) AEs—and any other issues of special concern outside South Africa should be submitted every six (6) months (using the progress report form in ZAF-18 ).
- An initial detailed report of new information impacting the risk-benefit profile of the IP or conduct of trial should be submitted within three (3) calendar days; a follow-up report should be submitted within an additional six (6) months.
- An initial detailed report of other major safety concerns (e.g., changes in nature, severity, or frequency of risk factors) should be submitted within 15 days of knowledge of the concern; a follow-up report should be submitted within an additional six (6) months.
- DSURs should be submitted within one year from approval of the study and annually thereafter.
In addition, SAHPRA reserves the right to impose additional reporting timelines on an individual protocol basis, and it may require expedited reporting of AEs of special interest, whether serious or not.
See the G-SafetyRpt for details on the contents of the reports and other safety report requirements.
Form Completion & Delivery Requirements
Per the G-SafetyRpt and ZAF-19 , the SAHPRA’s Safety Reporting During Clinical Trials Form ( ZAF-19 ) should be used to complete SAE/ADR reports—for both initial and follow-up safety reports. The G-SafetyRpt indicates that adverse drug reactions occurring during post-marketing studies (Phase 4 and observational studies) should be reported to the Vigilance Unit of SAHPRA, and adverse drug reactions occurring during the use of concomitant and/or comparator medicine in a clinical trial should be reported to the Clinical Trial Unit of SAHPRA. Reportable safety information must be sent to:
- [email protected] for clinical trials (per ZAF-47 , [email protected] should be used for individual patient SAEs and related queries)
- [email protected] or [email protected] for post-marketing studies
- [email protected] for reporting of SAEs for medicines used under Section 21
As per ZAF-47 , the following is the contact information for pharmacovigilance-related submissions:
- ADR reports in an e2b in an xml format: [email protected]
- All other ADR reports: [email protected]
- Pharmacovigilance related queries: [email protected]
- Documentation relating to identified safety concerns, responses to recommendations, and Risk Management Plans (RMPs): [email protected]
G-CTA-Electronic details the requirements for electronic submission of individual SAEs. All SAEs should be submitted to [email protected] with a cover letter detailing:
- The title of the study
- The SAHPRA reference number
- Protocol number
- Name of site
- Patient study ID
- Cause of SAE
- Causality and SAE reporting form
- Other applicable information
The email subject line should include the following information: SAE, protocol number, and SAHPRA database tracking number.
Interim and Annual Progress Reports
In accordance with the GRMRSA , the person authorized by the South African Health Products Regulatory Authority (SAHPRA) to conduct a clinical trial (i.e., the sponsor) must submit progress reports to the SAHPRA every six (6) months from the application approval date. The SA-GCPs requires the investigator to submit written progress reports to the ethics committee (EC) annually and to SAHPRA every six (6) months. ECs and SAHPRA may request reports more frequently.
Per the GRMRSA , the SA-GCPs , and G-SafetyRpt , the six-month report ( ZAF-18 ) must include the following (Note: the sources provide overlapping and unique elements so each of the items listed below will not necessarily be in each source):
- SAHPRA database tracking number
- Study title
- Details of the sponsor
- Progress to date or the outcome in case of completed research
- Whether participant follow up is still active or has been completed
- List of all active trial sites, addresses, and principal investigators (PIs)
- Trial information, including date of approval of study, treatment hold (if applicable), and expected date of completion
- Number of participants per site and current enrollment status
- Sponsor comment on progress to date
- Summary Data Safety Monitoring Board or Safety Committee recommendations and relevant safety data
- Serious adverse events and suspected unexpected serious adverse reactions for all participants per site in South Africa, including identification of previous safety reports submitted to SAHPRA concerning a similar suspected adverse reaction and an analysis of their connection
- Any safety issues of special concern outside of South Africa
- Line listing of all critical and major protocol violations/noncompliance and resolutions/actions taken at a site or conditions of approval
- Principal investigator (PI) comment on other major safety concerns
- Signature of the PI
- Signature of the sponsor
Note that the SA-GCPs directs the investigator to promptly provide written reports to the sponsor/applicant, the EC, and where applicable, the institution on changes that significantly affect trial conduct and/or increase the risk of participant harm.
Final Report
The sponsor is required to submit a final progress report to the SAHPRA 30 days following the trial’s completion as stated in the GRMRSA and the G-SafetyRpt . Further, per G-SafetyRpt , a final study report should be submitted within 180 days of clinical trial completion or termination.
In addition, per the SA-GCPs , upon the trial’s end, the investigator must inform the institution (if applicable), the EC, and SAHPRA and provide them with a summary of the trial outcome and other required reports.
The SA-GCPs specifies that the sponsor must ensure that trial results and outcomes are reported to the investigators, SAHPRA, and the National Department of Health (NDOH) via the South African National Clinical Trials Register (SANCTR) ( ZAF-48 ) within one (1) year of the study’s completion. The sponsor and the PI are responsible for appropriate dissemination of the trial findings.
As defined in the SA-GCPs , a sponsor is the person or organization responsible for the initiation, management, or financing of a clinical trial. A sponsor can be a pharmaceutical company, the principal investigator (PI), a funding body, or an individual or organization designated by the funding body or academic institution. An applicant can be an individual, company, institution, or organization that acts on behalf of the sponsor to initiate and manage the trial as its local representative. In the case of an international sponsor, a local applicant designated by the sponsor is responsible for initiation and management of the trial in the local context.
Per the SA-GCPs , a sponsor may transfer any or all trial-related duties and functions to a contract research organization (CRO). However, the sponsor is always ultimately responsible for the study data quality and integrity. Further, per the G-Monitor , the sponsor is solely responsible for adequate oversight of clinical trial conduct, including the justification for and selection of monitoring methods. Any trial-related responsibilities transferred to and assumed by a CRO should be specified in writing. The sponsor retains those responsibilities not specifically transferred to and assumed by a CRO.
As set forth in the SA-GCPs , the sponsor is responsible for using qualified individuals (e.g., biostatisticians, clinical pharmacologists, and physicians), as appropriate, throughout all stages of the trial process. Sponsors should select investigator(s) who are qualified by training and experience and have adequate resources to conduct the proposed clinical trial. Further, per the G-Monitor , the sponsor should consider previous experience with the investigator or site, workload of the investigator, and resource availability at the study site during investigator and site selection. Per the G-Capacity , clinical trial applications should include evidence and activity plans to build capacity at each study site as well as enhancing research activities and skills of professionals from historically disadvantaged groups. Mandatory training in Good Clinical Practice (GCP) forms a part of capacity building. To support transformation and capacity building, the South African Health Products Regulatory Authority (SAHPRA) states that the sponsor must have a policy on “Capacity Building and Transformation in Clinical Research in SA” in place, and preferentially select sites that are compliant. See G-Capacity , for detailed information on actions that will comply with this requirement.
According to the SA-GCPs , the sponsor must also define and allocate all study related duties and responsibilities to the investigator prior to initiating the study.
In addition, per ZAF-21 , to add or change investigators and/or additional sites to an approved clinical trial, the sponsor must submit a signed application to SAHPRA. See ZAF-21 for details.
Per the G-CTInvestigators , SAHPRA will recognize and approve categories of investigators for trial leadership. The principal investigator (PI) must be a South Africa-based scientist, who has sole or joint responsibility for the design, conduct, and delegation of trial responsibilities, analysis, and reporting. The PI is accountable to the sponsor and regulatory authorities. The PI can designate and supervise sub-principal investigator(s) (Sub-PI) of which at least one (1) must be a clinician and registered with the appropriate statutory entity to provide clinical oversight within their scope of practice. Further, the SAHPRA recognizes a category of co-principal investigator (co-PI), which allows for a team consisting of two (2) co-PIs to lead a study at a site. At least one (1) of the co-PIs must be a clinician registered with the appropriate statutory body and qualified to provide clinical oversight within their scope of practice. For multi-center studies, there must be a national PI appointed, who may or may not be a site PI. The national PI must have appropriate experience and expertise in that field and must be responsible for the application to the SAHPRA to conduct the study. The national PI must meet all other requirements to be a PI and sign a declaration accepting the responsibility as national PI and sign off on the clinical trial application. For more information on PI requirements, roles, and responsibilities, see the G-CTInvestigators .
Per the SA-GCPs , all parties involved in the conduct of a trial should be familiar with the guidance in the International Council for Harmonisation's Guideline for Good Clinical Practice E6(R2) ( ZAF-27 ) and other international guidelines. Additionally, the investigator must agree to conduct the trial in compliance with the SA-GCPs , ZAF-27 , SAHPRA requirements, and the ethics committee approved protocol. In the event of an interpretation conflict between the SA-GCPs and an international guideline, the SA-GCPs take precedence.
Foreign Sponsor Responsibilities
As required in the SA-GCPs , if South Africa is selected as a clinical trial site but the country of origin or other high-income countries are not, the sponsor must explain the reason(s) why and provide a clear ethical justification. Further, multi-national trials should ensure that a reasonable proportion of project team members are South African researchers, including scientists and health care professionals and those from previously disadvantaged backgrounds.
Data and Safety Monitoring Board
Per the SA-GCPs , the sponsor may establish an independent Data Safety Monitoring Board (DSMB) to assess the progress of a clinical trial, including safety data and critical efficacy endpoints at intervals, and to recommend to the sponsor whether to continue, modify, or stop a trial. The DSMB must have written standard operating procedures and must maintain written records of all its meetings.
Multicenter Studies
Per the SA-GCPs , if the trial is a multicenter and/or multi-country trial, any differences in trial designs between the South African and other sites must be clearly documented and explained in the trial protocol and/or related documents. In addition, international research groups must comply with South African regulatory requirements, and researchers must adapt the trial design and informed consent procedures to take into account local conditions and characteristics.
As set forth in the G-Insurance and the SA-GCPs , all clinical trial sponsors and investigators must obtain adequate insurance and indemnity to cover any liability claims during the conduct of a clinical trial, in accordance with the responsibilities described in the SA-GCPs . As delineated in the SA-GCPs and G-Insurance , a sponsor must follow the principles set forth in the Association of the British Pharmaceutical Industry’s (ABPI) guidelines ( ZAF-26 and ZAF-25 ) to comply with South Africa’s clinical trial insurance requirements. Per the SA-GCPs , research participants should not bear any financial cost to rectify harms that occur as a result of trial participation. The insurer pays the medical costs of necessary treatment to restore the previous position of the participant, if possible, when bodily or other injury is attributable to trial participation. Only bodily injuries of an enduring and disabling character (including exacerbation of an existing condition) and/or death are covered by the insurance. Temporary pain or discomfort or less serious or curable complaints are generally not regarded as trial-related, bodily injury. In the case of an in-utero injury due to the mother’s participation, payment for medical expenses proceeds as though the unborn child is a research participant. For additional details on limitations on liability, dispute resolution, weighting of risk factors, and insurance settlements, see the SA-GCPs .
Per the G-Insurance , the application to conduct a clinical trial must include evidence of comprehensive no fault insurance for serious injury and harm and/or death. In addition, the sponsor must provide indemnification for all investigators and trial sites involved in their clinical studies on compliance with the protocol requirements. In cases where the investigators/site staff were negligent and/or did not comply with the protocol requirements, personal malpractice insurance would apply.
As delineated in the G-Insurance and ZAF-23 , an insurance certificate and indemnity must be included in the clinical trial application submitted to the South African Health Products Regulatory Authority (SAHPRA) . Per the G-Insurance , the sponsor must include details of the insurance, including the following:
- Name and local address of the insurance company, including contact name and telephone number
- Title and protocol number of the clinical trial
- Date of commencement and termination of coverage
- Liability limit – per occurrence and total per occurrence and total for the study. Note that the limit should be adequate enough to cover extended stay in an intensive care unit or hospital
- Date of issuance of the insurance policy and expiry thereof
- Original or electronic signature of the insurer
- Special conditions if any. It is unacceptable to have special conditions which may invalidate or abate the clinical trial cover
- Any additional coverage
- Declaration of compliance with the SA-GCPs and ABPI guidelines on the certificate and in the patient information leaflet
- Where the insurance is not provided by a local company, a local insurance vendor must be identified with full details
- Insurance policy number
- The amount insured
Compensation
Injury or Death
As set forth in the G-Insurance , all clinical trial sponsors and investigators must have adequate insurance to cover any liability claims during the conduct of a clinical trial, in accordance with the responsibilities as described in the SA-GCPs . As delineated in the SA-GCPs and G-Insurance , a sponsor must follow the principles set forth in the ABPI guidelines ( ZAF-26 and ZAF-25 ) to comply with South Africa’s participant compensation and treatment requirements for trial-related injuries. The guidelines state that the sponsor should furnish written assurance to the investigator that the sponsor will agree to pay compensation to participants and/or their legal heirs in the event of trial-related injuries or death. The investigator, in turn, communicates this information to the relevant ethics committee (EC).
The SA-GCPs , the G-Insurance , and ZAF-26 provide several compensation principles to guide sponsors in fulfilling their obligations (Note: the sources provide overlapping and unique elements so each of the items listed below will not necessarily be in each source):
- Compensation should be paid when it can be demonstrated that a causal relationship exists between a participant’s injury and their participation in a trial
- Compensation should be paid when the injury results in permanent injury or disability to the participant
- When there is an adverse reaction to a medicinal product under trial, and injury is caused by a procedure adopted to deal with that adverse reaction
- The sponsor/applicant is under strict liability with respect to injuries caused by the investigational product (IP), and research participants should not bear any financial cost to rectify harms that occur as a result of trial participation
- The insurer should pay the medical costs of necessary treatment to restore the previous position of the participant, if possible
- In the case of an in-utero injury due to the mother’s participation, payment for medical expenses proceeds as though the unborn child is a research participant
- In principle, only bodily injuries of an enduring and disabling character (including exacerbation of an existing condition) and/or death are covered by the insurance; temporary pain or discomfort or less serious or curable complaints are generally not regarded as trial-related, bodily injury
- Where there is an adverse reaction to an IP and the injury is caused by a procedure adopted to deal with that adverse reaction, compensation should be paid for such injury as if it were caused directly by the IP
- Payment for medical expenses is made without acknowledgement of any legal liability and is thus to be understood to be an ex-gratia payment
- The provision of insurance cover and payment of medical expenses does not mean that an injured participant may not pursue legal action against the sponsor for loss or harm not covered by the insurance; however, an argument that pain and suffering, loss of income, and other possible claims should be paid for by the sponsor’s insurer is not sound in South African law and will not succeed
- The likelihood of an adverse reaction, or the fact that the participant has freely consented (whether in writing or otherwise) to participate in the trial should not exclude the participant from being eligible for compensation
According to the SA-GCPs and ZAF-26 , the amount of compensation to be paid to the participant should be appropriate to the nature, severity, and persistence of the injury. The compensation should also be generally consistent with the amount of damages commonly awarded for similar injuries. The amount paid in compensation should be abated, or in certain circumstances excluded, in light of the following factors (which will depend on the risk level the participant can reasonably be expected to accept):
- The seriousness of the disease being treated
- The degree of probability that adverse reactions will occur and any warning given
- The risks and benefits of the established treatments relative to those known or suspected of the trial medicines
ZAF-26 provides that in any case where the sponsor agrees to pay the participant, but the two (2) parties differ on what is the appropriate level of compensation, it is recommended that the sponsor agree to seek, at the sponsor’s own cost, the opinion of a mutually acceptable independent expert. This opinion should then be made available to the participant(s), and the expert’s opinion should be given substantial weight by the sponsor in reaching a decision on the payment amount.
Additionally, any participant claims pursuant to ZAF-26 should be made to the sponsor, preferably via the investigator. The participant should include details on the nature and background of the claim, which the sponsor should review expeditiously. The review process may be delayed if the participant requests an authority to examine any medical records relevant to the claim.
Trial Participation
As specified in the G-TIECompensation and the SA-GCPs , the sponsor or the designated representative is responsible for providing compensation to research participants. The SA-GCPs state that before the clinical phase of the trial commences, the EC must approve the documentation on participant compensation. Per the G-EthicsHR-ZAF , the SA-GCPs , and the G-TIECompensation , compensation should be based on time, inconvenience, and expenses. In addition, the G-EthicsHR-ZAF and the SA-GCPs also address researcher requirements to budget for participant travel and other expenses. (See the G-EthicsHR-ZAF for detailed information). See ZAF-5 for an analysis of ethical considerations regarding payment of trial participants in South Africa.
The G-TIECompensation guides sponsors of approved clinical trials and proposes a model for minimum compensation that can be paid. It is not intended as an exclusive approach and the SAHPRA reserves the right to request any additional information. In addition, G-TIECompensation is not applicable to Phase I clinical trials, which pose a higher risk for participants and should be compensated on a different scale.
Post-Trial Access
The G-PostCTAccess guides sponsors on when to consider post-trial or continued access (PTA/CA) to the IP following the trial’s conclusion. Only those participants who derive benefit from the IP will be considered (this excludes participants on standard of care, placebo, and registered medicines). Where appropriate and available, the possibility of PTA/CA should be disclosed to and discussed with potential participants during the initial informed consent process or via a separate consent process. Where appropriate and/or available, details of potential PTA/CA should be included in the clinical trial application form, informed consent form, and patient information leaflet. Additional considerations include the following:
- PTA/CA is not applicable for Phase I and II studies. However, PTA/CA may be necessary for particular diseases (e.g., cancer or rare diseases).
- PTA/CA should be considered for Phase III studies when there is no registered and marketed standard of care in South Africa, provided that data from interim or final analyses shows that access is clinically justifiable.
- PTA/CA is not applicable to Phase IV studies
- A minimum of four (4) years after completion of the study is recommended as the acceptable time period to provide PTA/CA to the participants, unless there are compelling reasons for determining otherwise.
- During the PTA/CA period, the sponsor must ensure monitoring and oversight of participants using the IP.
Quality Assurance/Quality Control
Per the SA-GCPs , the sponsor is responsible for implementing a quality management system to manage quality throughout the design, conduct, recording, evaluation, reporting, and archiving of clinical trials. This quality management system should adopt a risk-based approach for risk identification, evaluation, control, communication, and reporting. The sponsor should focus on trial activities that promote human participant protection and reliability of trial results, which include using qualified individuals, designating qualified medical personnel to respond to trial-related medical questions, and ensuring all aspects of the trial are operationally feasible and avoiding unnecessary complexity, procedures, and data collection. With respect to quality assurance (QA) and quality control (QC), the sponsor is responsible for implementing and maintaining QA and QC systems with written standard operating procedures (SOPs) to ensure that trials are conducted and data are generated, documented (recorded), and reported in compliance with the protocol, good clinical practice, and the applicable regulatory requirement(s).
Per the G-Monitor , the responsibility for adequate oversight of the conduct of a clinical trial, including the justification for and selection of monitoring methods, remains that of the sponsor solely.
Per the SA-GCPs , all parties involved in the conduct of a trial should be familiar with guidance in the International Council for Harmonisation's Guideline for Good Clinical Practice E6(R2) ( ZAF-27 ) and other international guidelines. Additionally, the investigator must agree to conduct the trial in compliance with the SA-GCPs , ZAF-27 , South African Health Products Regulatory Authority (SAHPRA) requirements, and the ethics committee (EC) approved protocol. In the event of an interpretation conflict between the SA-GCPs and an international guideline, the SA-GCPs take precedence.
Monitoring Requirements
In accordance with the SA-GCPs , the sponsor must conduct an independent audit to evaluate trial conduct and compliance with the protocol, procedures, good clinical practice, and the applicable regulatory requirements. The sponsor must appoint individuals who are independent of the clinical trials to conduct the audits and ensure that the auditors are qualified by training and experience to conduct audits properly. The sponsor's audit plan and procedures for a trial audit must be guided by the number of participants in the trial, the type and complexity of the trial, the level of risks to the trial participants, and any identified problem(s). Observations and findings of the auditors must be documented. The sponsor is responsible for obtaining agreement from all involved parties to ensure direct access to all trial related sites, source data/documents, and reports for monitoring and auditing purposes, and inspection by domestic and foreign regulatory authorities.
In addition, per the G-Monitor , the sponsor’s monitoring plan should include planned audits to ensure that monitoring activities are in accordance with the monitoring plan, applicable regulations, guidance, and sponsor’s plans and policies.
Premature Study Termination/Suspension
Per the SA-GCPs , if a trial is prematurely terminated or suspended for any reason, the investigator must promptly inform the trial participants and ensure appropriate therapy and follow-up for them. If the investigator, sponsor, institution, SAHPRA, or the EC terminate or suspend a trial, the investigator must promptly inform the other parties with a detailed written explanation for the termination or suspension. The sponsor is also responsible for ensuring that the South African National Clinical Trials Register (SANCTR) ( ZAF-48 ) is updated as well.
Electronic Data Processing System
Per the SA-GCPs , the sponsor must ensure that the electronic data processing system conforms to the specific documented requirements for completeness, accuracy, reliability, and consistency of intended performance, and that standard operating procedures for using these systems are maintained. In addition, the sponsor must:
- Ensure that the systems are designed to document data changes without deleting previously entered data (i.e., maintain an audit trail)
- Maintain a security system that prevents unauthorized access to the data
- Maintain a register of persons authorized to make data changes
- Maintain adequate data backup
- Ensure that blinding, if any, is maintained during data entry and processing
- Ensure the integrity and confidentiality of data, including any that describe the context, content, and structure of the data – especially when making changes to computerized systems
- If data are transformed during processing, it must be possible to compare the original data and observations with the processed data
- Use an unambiguous participant identification code that allows identification of all data reported for each participant
- Report any transfer of ownership of the data to South African Health Products Regulatory Authority (SAHPRA)
Per the G-Monitor , when developing a study’s monitoring plan, the sponsor should consider how it uses electronic data capture (EDC) systems. EDC systems that are capable of assessing quality metrics in real time will help identify high-risk sites that need more intensive monitoring.
Records Management
As set forth in the SA-GCPs , the sponsor should inform the investigator(s) in writing of the need for record retention, and should notify these parties in writing when the trial related records are no longer needed. The sponsor, or other data owners, must retain all the sponsor-specific essential documents pertaining to the trial for not less than 10 years or until at least two (2) years have elapsed since the formal discontinuation of clinical development of the investigational product (IP).
Responsible Parties
For the purposes of data protection requirements, the POPIA provides that the “responsible party” is a public or private body or any other person that, alone or in conjunction with others, determines the purpose of and means for processing personal information.
Data Protection
Per the POPIA , participants have the right to privacy, which includes a right to protection against the unlawful collection, retention, dissemination, and use of personal information by public and private bodies. This right to privacy is subject to justifiable limitations that are aimed at protecting other rights and interests (e.g., the right of access to information). Additional information on the rights of data subjects is provided in the POPIA .
The POPIA states that the responsible party must protect the constitutional right to privacy by safeguarding personal information when it is processed. The law provides conditions under which personal information may be gathered and processed.
- Accountability – The responsible party must ensure that the conditions and all the measures in the POPIA are complied with at the time of the purpose and means of processing is determined
- Processing limitation – Personal information may only be processed in a fair and lawful manner and only with the consent of the data subject
- Purpose specification – Personal information may only be processed for specific, explicitly defined, and legitimate reasons
- Further processing limitation – Personal information may not be processed for a secondary purpose unless that processing is compatible with the original purpose
- Information quality – The responsible party must take reasonable steps to ensure that the personal information collected is complete, accurate, not misleading, and updated where necessary
- Openness – The data subject whose information you are collecting must be aware that you are collecting such personal information and for what purpose the information will be used
- Security safeguards – Personal information must be kept secure against the risk of loss, unlawful access, interference, modification, unauthorized destruction and disclosure
- Data subject participation – Data subjects may request whether their personal information is held, as well as the correction and/or deletion of any personal information held about them
The POPIA establishes a duty requiring a public or private body to register its Information Officer with the Information Regulator (South Africa) . Per the POPIA , the Information Officer is responsible for compliance with lawful processing of information and working with and responding to requests by the Regulator. Per the POPIA-Regs , the Information Officer has further responsibilities to:
- Develop, implement, monitor, and maintain a compliance framework
- Conduct a personal information impact assessment to ensure compliance with the conditions for the lawful processing of personal information
- Develop, monitor, and maintain a manual; and make it available upon request by any person, provide copies of the manual to any person upon request and payment of a fee to be determined by the Information Regulator from time to time
- Develop internal measures and systems to process requests for information or access
- Conduct internal awareness sessions on protection of personal information requirements
- Provide reasonable assistance free of charge to the data subject in objecting to processing of personal information (using Form 1 in the POPIA-Regs ) and/or correcting or revising a record of personal information (using Form 2 in the POPIA-Regs )
The POPIA provides that records of personal information for research may be retained longer than is necessary for achieving the purpose for which the information was collected or processed if the responsible party has established appropriate safeguards against the records being used for any other purposes.
For additional guidance on processing personal data, including guidance on “special personal information” (e.g., health history) and personal information of children, see the Information Regulator website.
Consent for Processing Personal Data
Per the POPIA and the POPIA-Regs , personal information may only be processed if the data subject and/or the legal representative(s) or guardian(s) consents to the processing. The responsible party bears the burden of proof for the consent. The data subject and/or the legal representative(s) or guardian(s) may withdraw consent at any time if the lawfulness of the processing of personal information will not be affected.
Obtaining Consent
In all South African clinical trials, a freely given, written informed consent is required to be obtained from each participant in accordance with the principles set forth in the NHA , the Declaration of Helsinki ( ZAF-44 ), the SA-GCPs , and the International Council for Harmonisation's Guideline for Good Clinical Practice E6(R2) ( ZAF-27 ).
As per the SA-GCPs , the G-EthicsHR-ZAF , and the G-GPHlthCare , the informed consent form (ICF) and patient information sheet(s) are essential documents that must be reviewed and approved by an accredited ethics committee (EC) based in South Africa and provided to the South African Health Products Regulatory Authority (SAHPRA) with the clinical trial application. (See the Required Elements section for details on what should be included in the form.) The principal investigator (PI), or a person designated by the PI, should provide research study information to the participant and/or the legal representative(s), or guardian(s). When drafting and presenting the ICF, special consideration must be taken with regard to the participant’s culture, traditional values, intelligence, and education. The informed consent document should be non-technical and understandable to the participant and in a participant’s preferred written language. The ICF content should be briefly and clearly presented, without coercion or unduly influencing a potential participant to enroll in the clinical trial.
The SA-GCPs directs that none of the oral or written information concerning the study, including the written ICF, should contain any language that causes the participant and/or the legal representative(s) or guardian(s) to waive or to appear to waive their legal rights, or that releases or appears to release the investigator(s), the institution, the sponsor, or the representatives from the sponsor’s liabilities for any negligence.
The G-GPHlthCare-IC states that the participant must be informed of any relevant new findings over the course of the study, and be given the choice to continue to participate or withdraw from the study. Per the SA-GCPs , written informed consent documentation and other participant-related information should be revised when new information that may be relevant to a participant’s consent or willingness to continue to participate in the trial becomes available. Any revisions must be submitted for ethics review and approval before implementation. Communication of the new information to participants must be documented.
Language Requirements
According to the SA-GCPs and the G-EthicsHR-ZAF , the ICF and any patient information sheet(s) should be written in English and in a vernacular language that the participant is able to understand. The G-GPHlthCare states that the researchers should provide information to the participants in a language that the participant understands and in a manner that takes into account the participant’s level of literacy, understanding, values, and personal belief systems.
Documenting Consent
As stated in the SA-GCPs , the G-EthicsHR-ZAF , and the G-GPHlthCare , the ICF should be signed by the participant and the PI, or the person designated by the PI. If the participant is incapable of giving an informed consent, the legal representative(s) or guardian(s) should sign the ICF. The original signed ICF and patient information sheet(s) should be retained by the investigator and a copy should be given to the participant. The SA-GCPs requires an additional copy of the signed ICF and a source document identifying the study and recording the participation dates should be placed in the participant’s medical records. According to the SA-GCPs , the G-EthicsHR-ZAF , the G-GPHlthCare , and the G-GPHlthCare-IC , in all cases, written informed consent must be obtained. Where the participant is illiterate and/or the legal representative(s) or guardian(s) is illiterate, verbal consent should be obtained in the presence of and countersigned by a literate witness. The participant and/or the participant’s legal representative(s) or guardian(s), the PI or person designated by the PI, and if applicable, a literate witness must personally sign the ICF. Further, the SA-GCPs states that the participant should indicate willingness to participate by making a mark (either a cross or a fingerprint). The witness signs to affirm that the participant willingly consented to participate. The witness dates the mark and signature.
Waiver of Consent
No information is currently available regarding conditions for waiving consent.
Based on the informed consent essential elements in the SA-GCPs , the G-EthicsHR-ZAF , the G-GPHlthCare , and the NHAParticipants , the informed consent form (ICF) should include the following statements or descriptions, as applicable (Note: the regulations provide overlapping and unique elements so each of the items listed below will not necessarily be in each source):
- The study involves research and an explanation of its nature and purpose
- The procedures to be followed
- Why the potential participant has been approached and their responsibilities
- The aspects of the clinical trial that are experimental
- Any foreseeable risks or discomforts to the participant, and when applicable, to an embryo, fetus, or nursing infant; information should include the probability and magnitude of the foreseeable risks of harm
- Any benefits to the participant or to others that may reasonably be expected from the research; if no benefit is expected, the participant should also be made aware of this
- A disclosure of appropriate alternative procedures or treatments, and their potential benefits and risks
- The probability for random assignment to each treatment
- Participation is voluntary, the participant may withdraw at any time, and refusal to participate will not involve any penalty or loss of benefits, or reduction in the level of care to which the participant is otherwise entitled
- Compensation and/or medical treatment available to the participant in the event of a trial-related injury
- The planned incentives, if any, to attract the participant and the planned reimbursements, if any, for time, inconvenience, and expenses
- The extent to which confidentiality of records identifying the participant will be maintained, the possibility of record access by the sponsor, the ethics committee (EC), or the South African Health Products Regulatory Authority (SAHPRA)
- EC contact details for information and concerns regarding the trial participants’ rights
- The sponsor’s identity
- Potential conflicts of interest of the principal investigator (PI)
- The consequences of a participant's decision to withdraw from the study
- Information about approval from the EC and SAHPRA
- The approximate number of participants in the research study, locally and globally
- The expected duration of participation
- An explanation of whom to contact in the event of research-related injury
- Foreseeable circumstances under which the investigator(s) may remove the participant without consent
- The participant and/or the legal representative(s) or guardian(s) will be notified if significant new findings developed during the study which may affect the participant's willingness to continue
See the Vulnerable Populations and Consent for Specimen sections for further information.
South Africa’s ethical standards promote respect for all human beings and safeguard the rights of research study participants. In accordance with the principles held forth in the Declaration of Helsinki ( ZAF-44 ), the SA-GCPs , the G-EthicsHR-ZAF , the G-GPHlthCare , the G-GPHlthCare-IC , the NHAParticipants , and the International Council for Harmonisation’s Guideline for Good Clinical Practice E6(R2) ( ZAF-27 ), a participant’s rights must be clearly addressed in the informed consent form (ICF) and during the informed consent process. Below are the basic rights for participants in clinical research studies. (See the Required Elements and Vulnerable Populations sections for additional information regarding requirements for participant rights.)
The Right to Participate, Abstain, or Withdraw
According to the NHA and the NHAParticipants , everyone has the right to participate in any decision affecting their health or treatment, including research. The participant and/or the legal representative(s) or guardian(s) should be informed that participation is voluntary, that the participant may withdraw from the research study at any time, and that refusal to participate will not involve any penalty or loss of benefits to which the participant is otherwise entitled.
The Right to Information
According to the G-GPHlthCare-IC , a potential research study participant has the right to be fully informed on the nature and purpose of the research study, its anticipated duration, the sponsor and investigator(s), any potential benefits or risks, study procedures, any compensation for participation, injury and/or treatment, and any significant new information regarding the research study. (See the Required Elements section for a more detailed list.)
Per POAIA , a participant may seek access to their clinical trial records, pursuant to their constitutional right of access to any information held by the State or by another person.
The Right to Privacy and Confidentiality
Per the G-GPHlthCare-IC , participants have the right to privacy and confidentiality, and the ICF must provide a statement identifying this right. It is the responsibility of the investigator to safeguard the confidentiality of research data to protect the identity and records of research participants.
The Right of Inquiry/Appeal
Per the G-GPHlthCare-IC , the research participant and/or the legal representative(s) or guardian(s) should be provided with contact information for the investigator(s), and the ethics committee to address clinical trial-related queries, in the event of any injury and/or to appeal against a violation of the participant’s rights. It is also required that the South African Health Products Regulatory Authority (SAHPRA) address and contact information be provided. (See the Required Elements section for more detailed information regarding participant rights.)
The Right to Safety and Welfare
The SA-GCPs and ZAF-44 clearly state that research participants have the right to safety and well-being, which must take precedence over the interest of science and society. The NHA and the NHAParticipants safeguard the rights of all South Africans including vulnerable populations.
The NHA and the G-EthicsHR-ZAF make provisions to protect the rights of a research participant during the informed consent process when the procedure is complicated by medical emergencies. As per the G-EthicsHR-ZAF , the ethics committee (EC) may approve a delay in obtaining informed consent for emergency medical research if:
- Inclusion in the trial is not contrary to the interests of the patient
- The research poses no more risk than is inherent to the participant’s condition, or would be caused by alternative treatments
- The participant, the participant’s next of kin, and/or legal representative(s) or guardian(s) will be informed as soon as is reasonably possible of the participant’s inclusion in the study, and have the option to withdraw from the study at any time
- The research is based on valid scientific hypotheses, and offers a realistic possibility of benefit over standard care
Per the G-CTAPHEmerg , the South African Health Products Regulatory Authority (SAHPRA) states that during a public health emergency, informed consent and the patient information sheet(s) remain essential documents that must be reviewed and approved by an EC and provided to the SAHPRA with the clinical trial application.
The NHA , the SA-GCPs , the G-EthicsHR-ZAF , the G-GPHlthCare , and the NHAParticipants require special considerations for vulnerable populations, and characterize them by limited education, limited economic resources, inadequate protection of human rights, discrimination due to health status, limited ability to provide informed consent, limited availability of health care and treatment options, or an inadequate understanding of scientific research. Vulnerable populations include children/minors, mentally and physically disabled, pregnant women, substance abusers, prisoners, armed forces, the homeless, the elderly, members of a group with a hierarchical structure, patients with incurable diseases, persons in nursing homes, unemployed or impoverished persons, patients in emergency situations, ethnic minority groups, nomads, refugees, and other vulnerable groups such as persons in dependent relationships.
The SA-GCPs state that ethics committees (ECs) must pay special attention to protecting participants from vulnerable populations. The ECs may impose additional measures such as imposing additional protective measures for the informed consent process or requiring increased monitoring and interim reporting on the participants’ welfare. As per the NHAParticipants , research with vulnerable participants must comply with the following requirements:
- Involve vulnerable persons only when non-vulnerable persons are not appropriate for inclusion
- Not systematically avoid inclusion of vulnerable participants because it is unfairly discriminatory, and would prevent this population from benefiting from relevant research
- Be responsive to health needs and priorities of vulnerable persons, and
- Provide special attention in the ethical review to ensure research-related risks are assessed and minimized, and appropriate consent procedures are followed
See the Children/Minors; Pregnant Women, Fetuses & Neonates; Prisoners ; and Mentally Impaired sections for additional information about these populations.
Persons in Dependent Relationships or Hierarchical Situations
As indicated in the SA-GCPs and the G-EthicsHR-ZAF , participants whose proposed involvement in research arises from dependent or hierarchical relationships need additional attention, and particular attention should be given to ensuring that their consent is both adequately informed and voluntary. In addition, per the NHAParticipants , research is appropriate when research-related risks of harm are minimized. These types of relationships include, but are not limited to, those who are in junior or subordinate positions in hierarchically structured groups, such as prisoners and prison authorities, older persons and their caregivers, and patients and healthcare professionals.
Persons Highly Dependent on Medical Care
Per G-EthicsHR-ZAF , participants who are highly dependent on medical care may have a limited capacity to provide informed consent due to the gravity of their medical condition. In addition, their medical condition may require invasive measures resulting in greater risk. There may also be a perception of coercion if a participant is reluctant to refuse consent for fear that it may compromise the medical treatment. The EC may approve a delay in obtaining informed consent for research participants highly dependent on medical care if the following conditions are met:
- Research is based on valid scientific hypotheses that support a reasonable possibility of more benefit than that offered by standard care
- Participation is not contrary to their medical interests
- Research interventions pose no more risk of harm than that inherent in the participant’s condition or alternative methods of treatment
- As soon as reasonably possible, the participant must be informed and give delayed consent and advised of the right to withdraw from the research without any reduction in quality of care
Persons with Physical Disabilities
As described in the G-EthicsHR-ZAF , recruitment strategies for research participation in general should be sensitive to the possibility that persons with physical disabilities may wish to volunteer and therefore should ensure that there are no unintended barriers to such participation (e.g., the absence of ramps or a lift for wheelchair-bound potential participants). Research involving participants with physical disabilities should anticipate possible barriers and include measures to minimize them.
Elderly Persons
As per the G-GPHlthCare , research involving elderly persons requires consent to be provided by the participant’s legal representative(s) or guardian(s) on that person's behalf. Because of their vulnerability, the elderly should not be included in research unless the research is necessary to promote the health of this population and unless this research cannot instead be performed on legally competent persons.
Research Involving Collectivities
Per the G-EthicsHR-ZAF , a collectivity is a distinct group characterized by common beliefs, values, social structures, and other features identifying them as a separate group. Investigators are required to obtain EC approval for research involving a collectivity when any of the following conditions apply:
- Property or information private to the group as a whole is studied or used
- Research requires the permission of people occupying positions of authority, or involves members acknowledged as representatives to participate
The SA-GCPs stipulate that minors are younger than 18 years old and are regarded as vulnerable persons due to their lack of legal capacity. The G-GPHlthCare-IC states that a person over the age of 18 years is an adult and is legally competent to decide on all forms of treatment and medical procedures. However, a child who is 12 years of age and older is legally competent to consent to a proposed investigation if the child is of sufficient maturity and is able to understand the benefits, risks, social, and other implications of the research. A minor's/child’s refusal to participate in research must be respected.
Per the SA-GCPs , documented permission from the legal representative(s) or guardian(s) must be obtained in advance prior to approaching the minor to request participation. According to the NHA , the G-EthicsHR-ZAF , the SA-GCPs , the G-GPHlthCare , and the G-GPHlthCare-IC , consent for minors/children to participate in research must be obtained from:
- The legal representative(s) or guardian(s) in all but exceptional circumstances (such as emergencies)
- The minor/child who is competent to make the decision
- Any organization or person required by law (defined in the NHA )
- Where the minor/child is not competent, assent from the minor/child and consent from the legal representative(s) and/or guardian(s)
According to the NHA , where research or experimentation is to be conducted on a minor for therapeutic purposes, the study may only be conducted when:
- It is in the best interests of the minor/child
- It is carried out in such manner and on such conditions as may be prescribed
- The consent of the minor’s parent or guardian is provided
Where research or experimentation is to be conducted on a minor for non-therapeutic purposes, the NHA , the NHAParticipants , the SA-GCPs , and the G-MinisterConsent state that a study may only be conducted when:
- The consent of the Minister of Health is provided, or, where appropriate, consent from a delegated authority
- The consent of the minor is provided when the minor is capable of understanding
See the NHAParticipants for detailed application requirements.
In addition, per the G-MinisterConsent , the Minister of Health may not give consent if any of the following circumstances apply:
- The study objective(s) can also be achieved if conducted on an adult
- The research is unlikely to significantly improve scientific understanding of the minor’s/child's condition, disease, or disorder to such an extent that it will result in significant benefit to the minor(s)/child(ren)
- The reasons for the consent to the research by the parent or guardian and, if applicable, the minor/child, are contrary to public policy
- The research poses a significant risk to the health of the minor
- The risk to the health or well-being of the minor is not significantly outweighed by the potential benefit
For more information on ministerial consent for non-therapeutic health research with minors, see the operational guidelines at the G-MinisterConsent .
As delineated in the G-EthicsHR-ZAF and the NHAParticipants , the following additional criteria must be met to conduct clinical trials with minors/children:
- The research study presents minimal risk
- The research study presents more than minimal risk, but potentially direct or anticipated benefit for the participant outweighs the risk
- The research presents more than minimal risk (minor increase), and may not have a direct benefit to the participant, but has a high probability of producing important and relevant information, and that benefit may outweigh the risk
- Adults are not appropriate participants for the research
In all cases, there should be sufficient reasons to justify why minors/children should be included as participants.
Assent Requirements
The SA-GCPs and the G-EthicsHR-ZAF require the ethics committee (EC) to ensure that adequate steps outlined in the clinical protocol are used to obtain a minor’s assent when, in the EC’s judgment, the minor is capable of providing such assent. When the EC determines that assent is required, it must also indicate whether and how such assent should be documented. A minor’s/child’s assent should not be assumed simply because of failure to object during the informed consent process. It is necessary for the minor/child and the legal representative(s) or guardian(s) to be in agreement on participation. The minor’s/child’s refusal to participate is final.
Per the POPIA , there is a general prohibition on the processing of personal information of children. However, a responsible party may process personal information concerning a child for research purposes to the extent that:
- The purpose serves a public interest, and the processing is necessary for the purpose; or
- It appears to be impossible or would involve a disproportionate effort to ask for consent, and sufficient guarantees are provided to ensure that the processing does not adversely affect the individual privacy of the child to a disproportionate extent.
As per the NHA and the G-EthicsHR-ZAF , any research studies involving pregnant women, women who may become pregnant, or fetuses, require additional safeguards to ensure the research conforms to appropriate ethical standards and upholds societal values. The ethics committee (EC) must provide particular attention to these participants due to the potential for additional health concerns that may arise during pregnancy, and the need to avoid unnecessary risk to the fetus.
The SA-GCPs stipulates that pregnant women, women planning to become pregnant, or breastfeeding women are usually excluded from human clinical trials where a new chemical entity (NCE) or medicines with no information on safety in pregnancy/lactation are investigated for treatment of a particular disease/condition or disorder. However, when safety and other relevant information is available, pregnant or breastfeeding women should be included in clinical trials to ensure that appropriate knowledge about NCEs for this group is developed.
According to the NHA , the G-EthicsHR-ZAF , and the NHAParticipants , a prisoner may not, even with consent, participate in any scientific experimentation, research study, or clinical trial except under limited conditions. Per the G-EthicsHR-ZAF , prisoners are considered a vulnerable class of persons because of the potential effect of incarceration on the voluntariness of the decision to participate in research. Neither coercion nor undue influence is acceptable in the informed consent process. Researchers should pay attention to whether their intended participants are prisoners who are awaiting trial or are convicted as different ethical issues arise for each group. The recruitment strategy design must pay careful attention to how coercion and undue influence will be avoided. Similarly, persons administering questionnaires or conducting interviews must be conscious of environmental factors that may influence voluntariness. The ethics committee (EC) should include, at least on an ad-hoc basis, a member with experience and knowledge of working with prisoners when deliberating on the protocol.
Per the G-EthicsHR-ZAF , research should be conducted on prisoners only if:
- Their participation is indispensable to the research
- The research cannot be conducted with non-prisoners
- The research concerns a problem of relevance to prisoners
- Sound informed consent processes can be ensured
- Engagement with relevant role players about the proposed research has occurred
Generally, it is unlikely that independent consent by minor prisoners will be justifiable.
According to the NHA , the SA-GCPs , the G-EthicsHR-ZAF , the G-GPHlthCare , and the NHAParticipants , sufficient justification must be provided for any research or treatment involving a participant who has a mental or intellectual impairment or substance abuse related disorder, and the research must be relevant to the mental disability or substance abuse disorder.
Per the G-EthicsHR-ZAF , research involving these populations must conform to the following requirements:
- The research, including observational research, is not contrary to the best interest of the participant
- The research, including observational research, places the incapacitated adult at no more than minimal risk
- The research involves greater than minimal risk but provides the prospect of direct benefit for the incapacitated adult; the degree of risk must be justified by the potential benefit
- The research, including observational research, involves greater than minimal risk, with no prospect of direct benefit to the incapacitated adult, but has a high probability of providing generalizable knowledge
- The legally appropriate person gives permission for the person to participate
- Where appropriate, the person will assent to participation (Note that the incapacitated person’s refusal or resistance to participate, as indicated by words or behavior, takes precedence over permission by a proxy)
The G-EthicsHR-ZAF and the G-GPHlthCare state that research involving unconscious persons requires consent to be provided by the participant’s legal representative(s) or guardian(s) on that person's behalf. Unconscious persons should not be included in research unless the research is necessary to promote the health of the population represented and unless this research cannot instead be performed on legally competent persons.
As delineated in the SA-GCPs and the PIC-S-GMP-Guide (which South Africa adopted pursuant to the SA-GMPs ), an investigational product is defined as a pharmaceutical form of an active ingredient or placebo being tested or used as a reference in a clinical trial. This includes:
- A product with a marketing authorization when used or assembled (formulated or packaged) in a different way from the approved form
- When used for an unapproved indication
- When used to gain further information about an approved use
Manufacturing
According to the SA-GMPs and the GRMRSA , the South African Health Products Regulatory Authority (SAHPRA) is responsible for authorizing the manufacture of investigational products (IPs) in South Africa. As delineated in the G-ManuImpExp , a manufacturer’s license for IPs is required for both total and partial manufacture, and for the various processes of dividing up, packaging, or presentation, in accordance with the MRSA . To obtain a license, the application form ( ZAF-55 ) should be emailed to SAHPRA at [email protected] , accompanied by the following information:
- Proof of payment
- Existing SAHPRA license for renewal and amendment applications
- Site Master File
- Signed declaration
- SAHPRA inspection resolution
- Intellectual property documentation
- Department of Health premises license
- Registration of responsible pharmacist
- South African Pharmacy Council (SAPC) Record of a Pharmacy
- SAPC Record of a Pharmacy Owner
- Municipal Approval/Zoning Certificate
Per ZAF-55 , the license is valid for five (5) years and the application to renew the license must be submitted at least 180 days before the expiration of the current license.
In addition, per ZAF-23 , a clinical trial application to SAHPRA must include a certificate of good manufacturing practice (GMP) for manufacture of the IP(s). The SA-GCPs also states that the sponsor must ensure that the IP (including active comparator and placebo, if applicable) is manufactured in accordance with applicable GMP standards.
Pursuant to the SA-GMPs , South Africa adopted the PIC-S-GMP-Guide for the manufacturing of therapeutic goods. The PIC-S-GMP-Guide includes requirements for a Certificate of Analysis to be issued by the manufacturer for all IPs to be used in a clinical trial. For GMP agreements with competent international regulatory authorities, the SA-GMPs states that these agreements do not permit automatic acceptance but may be used to enhance regulatory oversight and compliance. SAHPRA may request additional documentation and/or schedule an inspection to ensure GMP compliance. The following conditions demonstrate GMP compliance:
- The site has been approved by a recognized regulatory authority (RA) within the previous three (3) years
- The dosage form of the IP within the application is within the same dosage form grouping as the dosage form approved by the RA
- The product type applied for is the same as the product type approved by the recognized RA
- The activities applied for by the applicant are the same activities that have been approved by the recognized regulator
The SA-GCPs states that IPs may be imported into South Africa only after approval of the protocol by SAHPRA. Samples of the IP to be imported before trial approval require a SAHPRA license under MRSA . The sponsor must ensure that the IP (including active comparator and placebo, if applicable) is manufactured in accordance with any applicable GMP standards. Per G-ManuImpExp to import an IP, the applicant must submit an application form ( ZAF-55 ) to SAHPRA.
Per the G-ImprtPorts , SAHPRA’s Regulatory Compliance Unit is responsible for ensuring that health products at ports of entry meet importation requirements under MRSA , including for IPs. Imported IPs must be accompanied by the certificate of registration that proves authorization under the MRSA .
Please note: South Africa is party to the Nagoya Protocol on Access and Benefit-sharing ( ZAF-8 ), which may have implications for studies of IPs developed using certain non-human genetic resources (e.g., plants, animals, and microbes). For more information, see ZAF-34 .
Investigator’s Brochure
In accordance with the SA-GCPs , the sponsor is responsible for ensuring an up-to-date Investigator’s Brochure (IB) is available to the investigator; investigators must provide it to the responsible ethics committee (EC). In the case of an investigator-sponsored trial, the sponsor-investigator must determine whether an IB is available from the commercial manufacturer.
The SA-GCPs states that the IB should contain the following sections, each with literature references where appropriate:
- Table of Contents
- Summary: A brief summary (preferably not exceeding two (2) pages) to highlight the significant physical, chemical, pharmaceutical, pharmacological, toxicological, pharmacokinetic, metabolic, and clinical information available that is relevant to the stage of clinical development of the investigational product (IP)
- A brief introductory statement with the chemical name (and generic and trade name for an approved product) of the IP, all active ingredients in the IP, its pharmacological class and expected position within this class (e.g., advantages), the rationale for conducting research with the IP, and the anticipated prophylactic, therapeutic, and/or diagnostic indications. Also include a description of the general approach to be followed in evaluating the IP.
- Physical, chemical, and pharmaceutical properties and formulation parameters
- Pre-clinical studies (pharmacology, pharmacokinetics, toxicology, and metabolism profiles)
- Effects of IP in humans (pharmacology, pharmacokinetics, metabolism, and pharmacodynamics; safety and efficacy; regulatory and postmarketing experiences)
- Summary of data and guidance for the investigator(s)
Quality Management
As defined in the SA-GCPs , the sponsor must ensure that IPs are manufactured in accordance with good manufacturing practices (GMPs), including the requirements in Annex 13 of the PIC-S-GMP-Guide (which South Africa adopted pursuant to the SA-GMPs ). (See Product Management section for additional information on IP supply, storage, and handling requirements). As indicated in ZAF-23 , the following information must be furnished in the clinical trial application:
- Whether the IP contains an active substance of chemical origin or of biological/biotechnological origin
- IP name(s) and details (e.g., formulation(s) and strength(s))
- Properties of the IP (e.g., mechanism of action)
- Summary of pre-clinical findings (e.g., laboratory, animal, toxicity, or mutagenicity)
- Summary of clinical findings
- Comparator product(s) name(s) and details
- Concomitant name(s) and details including rescue medications
- Registration status of IP, concomitant, and/or comparator medicine(s); include the IB, South African Health Products Regulatory Authority (SAHPRA) -approved principal investigator (PI), and other international professional information (package inserts) if not approved in South Africa, and a Certificate of Analysis (CoA)
- Whether the IP is modified in relation to its original registration for the purpose of the clinical trial
- Estimated quantity of trial material (each drug detailed separately) for which exemption will be required, including for concomitant medicines to be imported
- Explanation for use of imported drugs when the same product is available in South Africa
- Details of receiving the drugs from supplier including storage, dispensing, and packaging of drugs
- Details of intention to register the IP or explain if registration is not envisioned
- Details of the manufacture, quality control, and stability of the IP (including IP destruction process) and include certificate of good manufacturing practice (GMP)
- Previous studies using this medicine that have been approved by SAHPRA, including the SAHPRA approval number, study title, protocol number, date of approval, national PI/PI, date(s) of progress report(s), and date of final report
See ZAF-23 for detailed instructions on IP submission requirements.
Per the PIC-S-GMP-Guide (which South Africa adopted pursuant to the SA-GMPs ), the release of IPs should not occur until after the authorized person has certified that the relevant requirements have been met. CoAs should be issued for each batch of intermediate or active pharmaceutical ingredient, on request. CoAs should be dated and signed by authorized personnel of the quality unit(s) and should show the name, address, and telephone number of the original manufacturer. See the PIC-S-GMP-Guide for certification requirements.
Investigational product (IP) labeling in South Africa must comply with the requirements set forth in the SA-GCPs , the GRMRSA , MRSA , and the PIC-S-GMP-Guide (which South Africa adopted pursuant to the SA-GMPs ). The GRMRSA states that for an IP to be used in a clinical trial, it must be properly labeled in English and at least one (1) other official language, and should appear in clearly legible and indelible letters. As set forth in the PIC-S-GMP-Guide , the following labeling information must be included on both the outer packaging and the immediate container:
- The name, address, and telephone number of the sponsor, contract research organization (CRO), or investigator
- The pharmaceutical dosage form, route of administration, quantity of dosage units, and in the case of open trials, the name/identifier and strength/potency
- The batch and/or code number to identify the contents and packaging operation
- A trial reference code allowing identification of the trial, site, investigator, and sponsor (if not given elsewhere)
- The trial participant identification number/treatment number and where relevant, the visit number
- The investigator name (if not already included above)
- Directions for use (reference may be made to a leaflet or other explanatory document intended for the trial participant or person administering the product)
- “For clinical trial use only” or similar wording
- The storage conditions
- The period of use (use-by date, expiration date, or re-test date as applicable), in month/year format and in a manner that avoids any ambiguity
- “Keep out of reach of children” except when the product is for use in trials where the product is not taken home by the participant
In addition, precautions against mislabeling should be intensified by trained staff (e.g., label reconciliation, line clearance, and in-process control checks by appropriately trained staff).
The SA-GCPs specify that in blinded trials, the IP should be coded and labeled in a manner that protects the blinding. The IP(s) coding system should include a mechanism that permits rapid IP(s) identification in case of a medical emergency but does not permit undetectable breaks of the blinding.
Supply, Storage, and Handling Requirements
As defined in the SA-GCPs , the sponsor is responsible for supplying a sufficient quantity of the investigational product (IP) after the sponsor obtains study approvals from the South African Health Products Regulatory Authority (SAHPRA) and the ethics committee (EC). The sponsor must ensure that written procedures include instructions and relevant documents for the investigator to follow for handling and storage of the IP for the trial. The procedures must address adequate and safe receipt, handling, storage, dispensing, retrieval of unused product from participants, and return of unused IP to the sponsor (or alternative disposition if authorized by the sponsor and in compliance with the SAHPRA-approved protocol). In addition, the sponsor must:
- Ensure timely delivery of the IP to the investigator
- Maintain records that document shipment, receipt, disposition, return, and destruction of the IP
- Maintain a system for retrieving the IP and then documenting such retrieval (e.g., for deficient product recall, reclaim after trial completion, and expired product reclaim)
- Maintain a system for disposal of unused IP and for its documentation
- Take steps to ensure that the IP is stable over the period of use
- Maintain sufficient quantities of the IP used in the trials to reconfirm specifications, if necessary, and maintain records of batch sample analyses and characteristics; to the extent that IP stability permits, samples should be retained until analyses of trial data are complete or as required by the applicable regulatory requirement(s), whichever is longer
- Provide and maintain a system for retrieving and disposing of trial-related waste (e.g., syringes and needles)
Per the SA-GCPs , the sponsor should determine acceptable temperatures, conditions, times for IP storage, reconstitution fluids/procedures, and devices for product infusion, if any, that comply with the SA-GPPs . The sponsor must inform all parties involved (e.g., monitors, investigators, pharmacists, storage managers) of these determinations.
The SA-GCPs specify that if significant formulation changes are made in the IP(s) or comparator product(s) during the course of clinical development, the results of any studies of the newly formulated product(s) should be made available prior to its use in the clinical trial. Refer to the SA-GCPs for detailed sponsor-related IP requirements.
Regarding packaging, the PIC-S-GMP-Guide indicates that IPs are normally packed individually for each participant in the clinical trial. The number of units to be packaged should be specified prior to the start of the packaging operations, including units necessary for carrying out quality control and any retention samples to be kept. Sufficient reconciliations should take place to ensure the correct quantity of each product required has been accounted for at each stage of processing. During packaging, the risk of product mix up must be minimized by using appropriate procedures and/or, specialized equipment as appropriate and relevant staff training. The packaging must ensure that the IP remains in good condition during transport and storage at intermediate destinations. Any opening or tampering of the outer packaging during transport should be readily discernible. Similarly, the SA-GCPs state that the IPs must be suitably packaged in a manner that will prevent contamination and unacceptable deterioration during transport and storage.
Record Requirements
Per the SA-GCPs , the sponsor, or other data owners, must retain all essential documents pertaining to the trial for not less than 10 years or until at least two (2) years have elapsed since the formal discontinuation of clinical development of the IP. In addition, the sponsor should obtain the investigator’s agreement to retain trial-related essential documents until the sponsor informs the investigator/institution that these documents are no longer needed.
In South Africa, the NHARegMicroLabs refers to a specimen as a “diagnostic specimen,” and defines it as any human or animal material, including excreta, secreta, blood and its components, tissue or tissue fluids, that is to be used for the purpose of diagnosis, but does not include live infected animals. The G-EthicsHR-ZAF , in turn, refers to a specimen as a “biological specimen,” and defines it as material from a person including blood and blood products, DNA, RNA, blastomeres, polar bodies, cultured cells, embryos, gametes, progenitor stem cells, small tissue biopsies, and growth factors.
The term “specimen” appears to be used interchangeably with “biological material” in South Africa. The NHABiol and the MTA-Human follow the G-EthicsHR-ZAF definition of biological specimen, defining “biological material” as material from a human being including DNA, RNA, blastomeres, polar bodies, cultured cells, embryos, gametes, progenitor stem cells, small tissue biopsies, and growth factors from the same. The G-EthicsHR-ZAF defines “human biological materials” with the same definition as is used for “biological specimen.”
In addition, the NHABloodCells generally refers to substances of human origin as biological substances.
Please refer to the G-EthicsHR-ZAF , the NHABiol , the NHA , the NHABloodCells , the NHATissue , and the NHAStemCell for more specific definitions of selected terms including blood, cultured cells, embryonic tissue, human tissue, plasma, stem cell, and genetic material.
Import/Export
Per the NHA , the MTA-Human , the NHABloodCells , the NHARegMicroLabs , the NHATissue , and the NHAStemCell , a permit must also be obtained from the National Department of Health (NDOH) Director General to import or export biological substances. Both the South African Health Products Regulatory Authority (SAHPRA) approval letter and the NDOH import/export permit must be included with each biological substance shipment. See also the Submission Content section for information on completing a clinical trial application.
As set forth in the NHA , the NHABloodCells , the NHARegMicroLabs , the NHATissue , and the NHAStemCell , the NDOH Director-General, as delegated by the NDOH Minister, is responsible for establishing regulations related to the import and export of biological substances. In addition, only the Minister can authorize an institution or hospital to import or export biological substances for research purposes.
In accordance with the NHA , the NHABloodCells , the NHARegMicroLabs , the NHATissue , and the NHAStemCell , the NDOH Director-General reviews and approves all import or export requests by an institution or hospital. These requests must be submitted in writing using the application forms that may be obtained by contacting the NDOH Permit Programme at [email protected] . The forms also appear as Annexures 1-6 in the NHABloodCells and Form 1 in the NHARegMicroLabs .
Upon review of the application, the Director-General will issue a permit or certificate authorizing the import or export request if the Director-General is satisfied that the submission meets the NHA , the NHABloodCells , the NHARegMicroLabs , the NHATissue , and the NHAStemCell requirements, as applicable. The permit will contain an expiration date for the approved biological substance(s).
General Import/Export Requirements for Biological Substances
The NHABloodCells states that each biological substance to be imported into South Africa must be accompanied by a certificate from the supplier stating that the substance has been exported in terms of the originating country’s applicable laws and regulations.
As per the NHABloodCells and ZAF-7 , export permits for biological substances may only be issued by the Director-General to a Southern African Development Community (SADC) member state or to a South African citizen, provided that the country’s market requirements have been met. An applicant must also be registered with the Health Professions Council of South Africa (HPCSA) and operating in South Africa in order to apply for a permit to import or export biological substances. The applicant must also provide the Director-General with written information on stock levels for this substance along with the export application.
Applicants to whom a permit has been issued must keep a record of the import or export and submit this information using the register forms listed in Annexures 4, 5, and 6 of the NHABloodCells . The forms must be submitted to the Director-General annually before the end of February, for the preceding calendar year.
Import/Export Requirements for Specific Biological Substance Categories
The NHABloodCells provides details on unique application requirements for specific types of biological substances as outlined below:
- Import of tissues being used for therapeutic purposes: application must be accompanied by donor health status
- Export of tissues or gametes: application must include written proof that the donated biological substance complies with the NHA requirements
- Import or export of placenta tissue, embryonic or fetal tissue, embryonic, fetal or umbilical stem cells: applications will only be approved with the Minister’s written consent
- Import or export of blood or blood products: applications must be accompanied by a national blood transfusion service certificate and test results. If no documentation is included, the applicant must submit a letter to the Director-General explaining the reason. The Director-General will decide whether tests must be conducted, and the Minister is authorized to determine whether the applicant’s institution can be exempted from these requirements.
Material Transfer Agreement
Per the MTA-Human , all the providers and recipients of human biological material for use in research or clinical trials under the auspices of ethics committees (ECs) must use the “Material Transfer Agreement of Human Biological Materials” in MTA-Human . The agreement must be signed by the research institution’s authorized representative and the EC. The EC’s obligations are to:
- Review and approve research proposals and protocols that require the transfer of human biological materials
- Review and approve the material transfer agreement and ensure it adequately safeguards human biological material and ethical requirements
- Review and approve all secondary use research if the material is to be transferred
The EC must be the last party to sign the agreement after all the provisions of MTA-Human have been satisfied.
In accordance with the NHA , the NHASpecAmend , the NHABiol , and the MTA-Human , prior to removing or withdrawing any biological material from the body of a living person for research purposes, consent must be obtained from that person in writing, before a competent witness. In the event that the person is a minor, the parents or guardians of that person must provide consent. Furthermore, when withdrawing blood, the NHASpecAmend requires written consent from persons older than 16 years. Per the MTA-Human , the sponsor must obtain the completed informed consent form (ICF) from the donors of human biological materials and data, and submit it with the project protocol to the ethics committee (EC) for approval. Further, the sponsor must submit the ICF for secondary uses of the material to the EC should the need arise. Secondary use is defined as the use of the materials for health research purposes other than the uses determined in the approved protocol.
The NHABiol specifically states that when taking biological samples from a child, where the person is younger than 18 years, Part 3, Section 129 of the ChildrensAct must be followed.
Additionally, the NHABiol requires the following consent for the removal or withdrawal of biological samples to treat a person with mental illness:
- The mentally ill person’s consent, if capable;
- A court appointed curator, spouse, next of kin, parent or guardian, major child, brother, or sister, partner or associate, if the mentally ill person is incapable of giving consent; and
- The head of the health institution in the case of an emergency
Similarly, the NHA and the NHABiol include consent provisions for the donation of human bodies and the tissue of deceased persons. These documents state that any person who is competent to make a will may donate their body or any specified tissue to be used after death for medical and dental purposes, as long as the person signs the will in the presence of at least two (2) competent witnesses. The person may also give consent to a post-mortem examination of their body for research purposes, and may select an institution or person as the recipient. In the absence of a donation as described above, the individual’s spouse, child over 18 years, parent, guardian, or brother/sister over 18 years may donate the person’s body or any specific tissue to an institution or person for research purposes. Please refer to the NHA and the NHABiol for detailed requirements. (See the Required Elements and Participant Rights sections for additional information on informed consent).
The SA-GCPs observe that many biological samples are collected during clinical interventions for diagnostic purposes, which means the potential for future use of these samples may be presented. Use of human biological materials and their associated data facilitates research in new technologies, which include genetic and genomic research, and cell and gene therapy (CGT). Sponsors and researchers should, therefore, follow the fundamental ethical principles that underpin all research involving human biological materials and their associated data. Research proposals must address specifically the social value of the research especially in the local context; how consent, privacy, confidentiality will be managed; and the potential effect on families, communities, and other groups. Specific concerns include protection of privacy and whether and how incidental findings are to be communicated to the person from whom the sample originates.
Human Tissue Sample Consent Requirements
The G-EthicsHR-ZAF presents separate consent provisions for the use of human tissue samples. With reference to human tissue samples, donor consent should be obtained where it is proposed to use tissue samples that have been held:
- in storage following, or in association with, clinical investigations
- in archives or banks, or removed during a clinical study, or used in research that may lead to harm, benefit, or injustice to a donor of such tissue
In addition, consent must be voluntary and specific to the purpose for which the tissue is to be used. The participant must be given full information about the study, be advised on storage and future use of samples, and be assured of data related confidentiality and privacy.
In addition, per the NHATissue , tissue banks are required to develop donor record management systems in which the tissue donor register contains the full identity and relationship of the consenting person. The system will also document tissue banking processes, including the process of obtaining informed written consent.
Human Stem Cell Consent Requirements
The NHAStemCell similarly states that authorized stem cell banks must retain a record of the donor’s written informed consent. Further, no person shall use stem cells or its therapeutic research products for educational purposes unless authorized by the National Department of Health (NDOH) and is compliant with the following requirements:
- Has obtained the donor’s informed written consent even in the case of residual tissue, blood, or blood products; and
- Is certain the donor has donated voluntarily, and it is properly documented
The NHA also indicates that the NDOH Minister may permit research on stem cells and zygotes that are not more than 14 days old on a written application, and if the applicant documents the research for record purposes, and prior consent is obtained from the donor.
See ZAF-3 for an analysis of human tissue legislation and stem cell therapy in South Africa.
Human Genetic Research Consent Requirements
The G-EthicsHR-ZAF states that the investigator or institution must obtain consent for human genetic research. Investigators and institutions must comply with numerous requirements to ensure participant consent, protection, and privacy rights are upheld with regard to the storage of genetic materials. See the G-EthicsHR-ZAF for consent requirement details.
In reference to both human tissue sample and genetic research consent, the G-EthicsHR-ZAF indicates that an EC may sometimes waive the consent requirement in cases where there is minimal risk of commercial exploitation or privacy violations. For additional details, see section 3.3 of the G-EthicsHR-ZAF .

Global Research
Global research in south africa.
View Larger Map
Despite being one of the most economically advanced African nations, South Africa continues to face many public health challenges. Tuberculosis (TB) is one of the leading causes of death in South Africa. According to the 2021 World Health Organization (WHO) Global TB Report, South Africa is ranked eighth among the 30 high-burden countries that contribute to 86% of the estimated incident TB cases globally. Additionally, South Africa remains the global epicenter of the HIV/AIDS epidemic. UNAIDS estimates that approximately 7.8 million people in South Africa are currently living with HIV. Drug-resistant malaria is also a growing health concern in the country, while schistosomiasis, endemic in rivers in the eastern part of the country, continues to have a debilitating impact on rural South African populations.
NIAID-Funded Activities
For more than 20 years, NIAID has collaborated with and supported many research projects and training initiatives with institutions in South Africa, mostly focused on HIV/AIDS and TB, with additional projects on other emerging and re-emerging infectious diseases. NIAID also supports a large number of research networks that have sites in South Africa.
Select NIAID-Supported Research in South Africa:
The u.s. – south africa program for collaborative biomedical research.
The U.S. – South Africa Program for Collaborative Biomedical Research was established through a Memorandum of Understanding between the South Africa Medical Research Council (SAMRC) and NIH in 2013. The program supports collaborations between U.S. and South African scientists on research related to TB, HIV, and HIV-associated comorbidities, including cancer and other infectious diseases. NIAID coordinates the trans-NIH engagement in this bilateral research program.
Regional Prospective Observational Research in Tuberculosis (RePORT) South Africa
RePORT South Africa is a jointly funded, multi-organizational, collaborative effort that supports regional TB research aimed at developing diagnostics and quantifying the impact of undiagnosed cases with the ultimate goal of improving outcomes in TB. RePORT South Africa has five clinical sites that recruits participants from six provinces across South Africa, including regions with extremely high incidences of TB and high rates of TB in people living with HIV. RePORT South Africa is a seminal member of RePORT International which consists of multiple regional RePORT networks to encourage coordinated, global TB/HIV research in biomarkers, diagnostics, prevention (vaccines) and treatment strategies. The consortium represents regional cohorts in Brazil, China, India, Indonesia, South Africa and the Philippines that are linked through the implementation of a common protocol and laboratory standards to promote interoperability for data sharing and specimen collection.
Scientific Advances
One-month tuberculosis prophylaxis as effective as nine-month regimen for people living with hiv.
Results from a NIAID-funded trial found that a one-month antibiotic regimen to prevent active TB was at least as safe and effective as the standard nine-month therapy for people living with HIV. 3000 adults and adolescents in South Africa and nine other countries who enrolled in the trial were more likely to complete the short-course regimen—consisting of daily doses of the antibiotics rifapentine and isoniazid for four weeks—than a nine-month regimen of daily isoniazid.
Outcomes of HIV-Positive-to-HIV-Positive Renal Transplantation
South Africa has a unique patient population of HIV-positive to HIV-positive organ transplant recipients. NIAID-supported investigators conducted a study to evaluate outcomes of renal transplantation in HIV-positive patients who received organs from HIV-positive donors and found favorable clinical outcomes and the absence of transmitted drug resistance support the use of HIV-positive-to-HIV-positive renal transplantation as a treatment option. Investigators found that superinfection is not a risk, and survival and graft loss rates are similar to HIV-negative donors.
Landmark TB Trial Identifies Shorter-Course Treatment Regimen
An international, randomized controlled trial found that a four-month daily regimen with high-dose rifapentine plus moxifloxacin is as safe and effective as the standard six-month daily regimen at curing drug-susceptible TB. The Phase 3, open-label trial, called Study 31/A5349, was led by the CDC’s Tuberculosis Trials Consortium (TBTC) with collaboration from the Advancing Clinical Therapeutics Globally for HIV/AIDS and Other Infections (ACTG) within DAIDS, NIAID. More than 2,500 participants enrolled at 34 clinical sites including South Africa, Kenya, Malawi, Uganda, and Zimbabwe. Shorter regimens enable patients to be cured faster and may reduce treatment costs, improve patient quality of life, increase therapy completion, and reduce development of drug resistance.
To conduct safe, high quality clinical research through International Good Clinical Practice. (GCP)
To advance local Healthcare through International Research
Centre of Clinical Research Excellence
Tiervlei Trial Centre (TTC) is a well-established private clinical trial centre, since 2000, specialising in the conduct of international and national clinical trials. We have successfully completed more than 500 clinical trials in several therapeutic areas and all phases of medicines development.
TTC has a dedicated operational team consisting of investigators, study coordinators, research nurses, pharmacists, lung- and laboratory technologists, administrators, project managers and recruitment specialists, all certified in GCP (Good Clinical Practice). Our team works in close collaboration with specialists in the private sector, as well as with local universities.
The pharmaceutical industry has recognized TTC as an International Site of Excellence
TTC offers a wide variety of services for our clients. Our services range from specialist consultations to training and development.

Patient Recruitment
TTC recognized the importance of patient recruitment in the ultimate success of a clinical trial. We have a multi-facetted…

Clinical Trial Service
TTC has a department for completion of study feasibility assessments within required time frame…

Special Investigations
Lung function tests, allergen skin Prick test and bronchial provocation tests are performed by qualified…

Medical Writing
TTC provides a professional medical writing service. Our team has extensive experience in Protocol and Statistical…
Specialist Consultation
TTC team provides specialist consultation to clinical trial participants. We work in collaboration with Specialists…

Collaboration with Universities
TTC provides a specialize clinical trial services to local universities ranging review of data on….

Collaboration with Government Hospitals
TTC recognizes the challenge facing the South African health system and provides equitable distribution of…

Get access to the latest in medical research & development
Ethical clinical trials.
At TTC we focus on providing our patients and investors with high-quality services. What sets us apart from other trial centres?
01. Transparency with patients
02. research to develop medicine, 03. international standard trial facilities, 04. our diversity of patients.
- Mon-Fri: 7 AM – 4 PM
- Saturday: Closed
- Sunday: Closed

Are You Interested In A Trial?
Contact us today to find out how you could benefit from being a trial patient and if you qualify
Meet our Doctors

Dr. Haylene Nell

Dr Maria Pretorius

Dr. Zarinah Mohamed

Dr. Karla Morrison

Dr. Jan Vermeulen

Dr Michiel M De Vries Basson

Dr Ramatsiele Petrus Masekoemeng

Dr. Taryn Ackerberg
Management team.

Sonnika Van Vuuren
Jan de bruin, find out more about our clinical trials.
Get more information about our trials from one of our friendly, qualified medical professionals and see how we can help you!
Subscribe to stay updated on all of our latest trials!
Subscribe I agree to the Privacy Policy .
I had a very complicated case and many clinics in the past refused to take me as a patient. Dr. Johnson however was not afraid to treat my unique tooth condition. What a great experience! Thank you Doctor and the Clinic!
Sandra Morrison
Friendly staff, welcoming atmosphere and the state-of-the-art interior make you relax and enjoy the process, so you don’t feel like you are in a hospital at all! Everything went so smoothly that I cannot even describe my happiness.
Jack Phillips
I have been scared of doctors my entire life, but my close friend recommended me the Clinic. I gave it a chance and was extremely surprised! Qualified doctors and individual approach. Highly recommended to all with any health issues!
021 957 9400
Subscribe to stay up to date on all the latest updates in the trials field.

Enter your search term
*Limited to most recent 250 articles Use advanced search to set an earlier date range
Sponsored by
Saving articles
Articles can be saved for quick future reference. This is a subscriber benefit. If you are already a subscriber, please log in to save this article. If you are not a subscriber, click on the View Subscription Options button to subscribe.
Article Saved
Contact us at [email protected]
Forgot Password
Please enter the email address that you used to subscribe on Engineering News. Your password will be sent to this address.
Content Restricted
This content is only available to subscribers
REAL ECONOMY NEWS
sponsored by
- LATEST NEWS
- LOADSHEDDING
- MULTIMEDIA LATEST VIDEOS REAL ECONOMY REPORTS SECOND TAKE AUDIO ARTICLES CREAMER MEDIA ON SAFM WEBINARS YOUTUBE
- SECTORS AGRICULTURE AUTOMOTIVE CHEMICALS CONSTRUCTION DEFENCE & AEROSPACE ECONOMY ELECTRICITY ENERGY ENVIRONMENTAL MANUFACTURING METALS MINING RENEWABLE ENERGY SERVICES TECHNOLOGY & COMMUNICATIONS TRADE TRANSPORT & LOGISTICS WATER
- SPONSORED POSTS
- ANNOUNCEMENTS
- BUSINESS THOUGHT LEADERSHIP
- MINING WEEKLY
- SHOWROOM PLUS
- PRODUCT PORTAL
- MADE IN SOUTH AFRICA
- PRESS OFFICE
- WEBINAR RECORDINGS
- COMPANY PROFILES
- VIRTUAL SHOWROOMS
- CREAMER MEDIA
- BACK COPIES
- BUSINESS LEADER
- SUPPLEMENTS
- FEATURES LIBRARY
- RESEARCH REPORTS
- PROJECT BROWSER
Article Enquiry
Research project launched into telemedicine and HIV/Aids management in South Africa
Email This Article
separate emails by commas, maximum limit of 4 addresses
A pharmacist dispensing HIV/Aids medication
Photo by Reuters

As a magazine-and-online subscriber to Creamer Media's Engineering News & Mining Weekly , you are entitled to one free research report of your choice . You would have received a promotional code at the time of your subscription. Have this code ready and click here . At the time of check-out, please enter your promotional code to download your free report. Email [email protected] if you have forgotten your promotional code. If you have previously accessed your free report, you can purchase additional Research Reports by clicking on the “Buy Report” button on this page. The most cost-effective way to access all our Research Reports is by subscribing to Creamer Media's Research Channel Africa - you can upgrade your subscription now at this link .
The most cost-effective way to access all our Research Reports is by subscribing to Creamer Media's Research Channel Africa - you can upgrade your subscription now at this link . For a full list of Research Channel Africa benefits, click here
If you are not a subscriber, you can either buy the individual research report by clicking on the ‘Buy Report’ button, or you can subscribe and, not only gain access to your one free report, but also enjoy all other subscriber benefits , including 1) an electronic archive of back issues of the weekly news magazine; 2) access to an industrial and mining projects browser; 3) access to a database of published articles; and 4) the ability to save articles for future reference. At the time of your subscription, Creamer Media’s subscriptions department will be in contact with you to ensure that you receive a copy of your preferred Research Report. The most cost-effective way to access all our Research Reports is by subscribing to Creamer Media's Research Channel Africa - you can upgrade your subscription now at this link .
If you are a Creamer Media subscriber, click here to log in.
30th May 2024
By: Rebecca Campbell
Creamer Media Senior Deputy Editor
Font size: - +

How effective is telemedicine as a differentiated service delivery model for the prevention of HIV/Aids in South Africa? This is the question to be addressed by a PhD researcher at Stellenbosch University’s Africa Centre for HIV/Aids Management, Rudi de Koker .
“Globally, there is a goal of ‘ending’ HIV/Aids by 2030,” he points out to Engineering News . “This will need the use of different service delivery models. One of these could be telemedicine. Frankly, our expectation is that telemedicine will indeed be valuable, but could it do more? How big a role could it, should it, play?”
Telemedicine – virtual consultations – has only been formalised in South Africa in recent years, having only been authorised by the Health Professions Council of South Africa (HPCSA) as an extraordinary measure required to address the Covid-19 pandemic. Officially, in South Africa, telemedicine is still only a temporary emergency measure.
Even under the impact of Covid, the HPCSA only authorised telemedicine for use in situations in which there was already an existing relationship between the patient and the healthcare provider. “Other than this, no single framework exists for the use of telemedicine in South Africa,” he notes. “If this research shows telemedicine is effective in this country, then we have the basis to justify the development of a national telemedicine framework, including a White Paper.”
Given that he is also project manager at Digital Health Cape Town (DHCT), De Koker’s interest in the topic is unsurprising. DHCT has developed and manages the HIV Clinicians Expert Telemedicine Platform, which is the largest HIV telemedicine platform in South Africa, covering the entire country (and not just Cape Town). Currently, the platform embraces 397 active trained pharmacists and 88 active trained nurses.
“The key research point, with regard to South Africa, is the use of telemedicine for virtual consultations to create access to treatment,” he stresses. “Also, because of the disparity between urban and rural areas in South Africa, I’d like to see how telemedicine can be utilised to link health seekers in rural areas closer to healthcare services. That’s my personal ambition.”
The research project should be completed by the end of 2026.
Edited by Creamer Media Reporter
Research Reports

Latest Multimedia

Latest News

ESAB South Arica, the leading supplier of high-end welding and cutting products to the Southern African industrial market is based in...

Supplier & Distributor of the Widest Range of Accurate & Easy-to-Use Alcohol Breathalysers
sponsored by

Press Office
Announcements
Subscribe to improve your user experience...
Option 1 (equivalent of R125 a month):
Receive a weekly copy of Creamer Media's Engineering News & Mining Weekly magazine (print copy for those in South Africa and e-magazine for those outside of South Africa) Receive daily email newsletters Access to full search results Access archive of magazine back copies Access to Projects in Progress Access to ONE Research Report of your choice in PDF format
Option 2 (equivalent of R375 a month):
All benefits from Option 1 PLUS Access to Creamer Media's Research Channel Africa for ALL Research Reports, in PDF format, on various industrial and mining sectors including Electricity; Water; Energy Transition; Hydrogen; Roads, Rail and Ports; Coal; Gold; Platinum; Battery Metals; etc.
Already a subscriber?
Forgotten your password?
MAGAZINE & ONLINE
R1500 (equivalent of R125 a month)
Receive weekly copy of Creamer Media's Engineering News & Mining Weekly magazine (print copy for those in South Africa and e-magazine for those outside of South Africa)
Access to full search results
Access archive of magazine back copies
Access to Projects in Progress
Access to ONE Research Report of your choice in PDF format
RESEARCH CHANNEL AFRICA
R4500 (equivalent of R375 a month)
All benefits from Option 1
Electricity
Energy Transition
Roads, Rail and Ports
Battery Metals
CORPORATE PACKAGES
Discounted prices based on volume
Receive all benefits from Option 1 or Option 2 delivered to numerous people at your company
Intranet integration access to all in your organisation


An official website of the United States government
Here’s how you know
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Contact Information
Laura Lewandowski, M.D., M.S., received her medical degree from Pennsylvania State University College of Medicine and her undergraduate degree from the College of the Holy Cross. She completed pediatric residency training at the University of Massachusetts Medical Center and went on to serve as a chief resident in pediatrics. Dr. Lewandowski completed a combined four-year pediatric rheumatology/global health fellowship at Duke University Medical Center, during which she characterized a pediatric lupus patient cohort in South Africa. She holds a Masters in Global Health from Duke University. During her fellowship, she established a registry of pediatric lupus patients that continues to grow and is now the largest cohort in sub-Saharan Africa. Dr. Lewandowski was awarded a Fogarty Global Health Fellowship and a Lupus Foundation Early Career award for her work with pediatric lupus patients in South Africa.
In 2015, she joined the NIAMS under Dr. Mariana Kaplan as a Lawrence Shulman Scholar in Translational Medicine. She currently holds the position of assistant clinical investigator and Head of the Lupus Genomics and Global Health Disparities Unit at the NIAMS (NIH).
Research Statement
The Lewandowski Lab seeks to use genomics and transcriptomics to further understand the pathogenesis of systemic lupus erythematosus (SLE). The lab's primary research focus is studying the genetic changes in early-onset SLE patients to understand important drivers of disease pathogenesis. By including diverse populations of pediatric SLE patients in our research studies, we hope to broaden the understanding of differences in disease manifestations and severity in patients of different ancestries around the world. Dr. Lewandowski also has an interest in advancing rheumatology care and research in lower resourced settings.
Dr. Lewandowski is a clinician who sees pediatric patients with SLE as part of the NIAMS Lupus Team at the NIH Clinical Center. She has held several leadership positions, including serving as co-chair of the Childhood Arthritis and Rheumatic Diseases Research Alliance (CARRA) Lupus Nephritis workgroup, co-chair of the CARRA Lupus Genetics Workgroup, chair of the CARRA Translation Research and Technology Committee Data Management, and chair of the CARRA Annual Meeting Planning Committee. Dr. Lewandowski is a member of the Early Career Investigator Committee within the American College of Rheumatology. Also, she has mentored students and trainees in North America and South Africa in rheumatology research. She is a USA representative on the Paediatric Musculoskeletal Task Force. She has published widely on lupus in Africa and the challenges to rheumatology clinical care, education, and practice in less-resourced countries.
Scientific Publications
Principles of pediatric lupus nephritis in a prospective contemporary multi-center cohort., tackling global challenges in pediatric rheumatology., global rheumatology in the time of covid-19., update on cardiovascular disease in lupus., survival in adults and children with systemic lupus erythematosus: a systematic review and bayesian meta-analysis of studies from 1950 to 2016., severe disease presentation and poor outcomes among pediatric systemic lupus erythematosus patients in south africa..
Penn State University College of Medicine, Hershey, PA M.D.
Duke University, Durham, NC M.S., Global Health
National Institutes of Health, Bethesda, MD Lawrence Shulman Scholar in Translational Research (2020)
Duke University Medical School, Durham, NC Pediatric Rheumatology Fellowship (2015)
Duke Global Health Institute, Durham, NC Global Health Fellowship (2015)
University of Massachusetts Medical School, Worcester, MA Chief Resident (2011)
University of Massachusetts Medical School, Worcester, MA Pediatric Residency (2010)
IMAGES
VIDEO
COMMENTS
WELCOME TO TASK TASK is a multinational, multi-site clinical research institute committed to improving global health through testing and progressing novelty medicines, vaccines and diagnostics in various therapeutic areas. Learn more about us Our Network.
The South African Clinical research association (SACRA) is an industry community association with the sole purpose of leading and serving as a conduit within the clinical trials community. SACRA is a non-profit organisation representing the clinical research industry in South Africa.
Who we are. The SAMRC was established in 1969 and is dedicated to improving the health of people in South Africa, through research, innovation, development, and technology transfer. The scope of research includes laboratory investigations, clinical research, and public health studies. We conduct research on South Africa's quadruple burden of ...
The South African National Clinical Trials Register (SANCTR) provides the public with updated information on clinical trials on human participants being conducted in South Africa. The Register provides you with information on a trials purpose; who can participate, where the trial is located, and contact details.
Clinical Research Institute of South Africa (CRISA), Proprietary Limited. The purpose of CRISA is to conduct clinical research in South Africa in collaboration with Department of Health on projects that lead to optimised care for patients and promote a public health approach to research and treatment. CRISA aims to integrate clinical research ...
Each clinical research site (CRS) brings a unique complement of skills, populations, facilities, and experience. We have conducted more than 210 research studies and clinical trials at 5 main Clinical Research Centres in Klerksdorp, Rustenburg, Tembisa, Pretoria and Mozambique. ... South Africa, 2193.
Research sites located in busy urban communities in three major SA provinces. CHRU has four Clinical Research Sites (CRS) in South Africa where it undertakes clinical trials. These are at: The Helen Joseph Hospital, Westdene, Johannesburg, Gauteng province. The Sizwe Tropical Disease Hospital, Sandringham, Johannesburg, Gauteng province.
We have been in the industry for over 20 Years. Established in 1999, OnQ Research has conducted more than 500 clinical trials in more than 10 African countries. Our clinical study sizes range from 1 to 3000 participants.
Clinical Research Investigator Site Management Organisation (CRISMO), is a Site Management Organisation established in 2015 with its head-office based in Germiston, South Africa. We bridge the capacity building gap in Africa by providing site management systems and operational support to research investigator sites in the conduct of clinical ...
LCRI is a dedicated clinical research facility founded by Dr Leon Fouché. We have conducted more than 80 Phase II, III, and IV clinical trials. With over 13 years of experience in medical investigation, we are elevating the standard of healthcare in our community as well as globally. ... South Africa +27 14 777 1939. News. Check out our latest ...
Conducting Clinical Trials in South Africa: A Revitalized Environment. South Africa was once a commonly considered country for inclusion in clinical research, but its popularity caused bottlenecks within its previously constructed regulatory structure. Simply put, the system became overwhelmed and could not keep up thus leading to long ...
The CRC is an early phase clinical trial unit equipped to conduct first-in-human studies for investigation medicinal products or investigation medical devices. The CRC has its own Research Pharmacy, as well as a Sample Processing Laboratory. A team of experienced staff can guide and advise researchers in key aspects of conducting their clinical ...
CRTC. The Clinical Trials Community Africa Network (CTCAN) is a network for clinical research stakeholders in Africa that seeks to enable increased, sustainable, and coordinated clinical trials on the continent. The network will build on the progress of the Clinical Trials Community (CTC) platform, which was created to increase the visibility ...
Together for the future of health. Find a clinical study for you. Welcome to Synexus, your dedicated Clinical Research Centre. We conduct clinical research studies with the help of volunteers like you. By registering with us, we hope that we can find a suitable study for you now, or in the future.
WHO WE ARE. Synergy Biomed Research Institute (SBRI), a diverse black-and-women-led organisation, was established in 2020 in East London, South Africa, to address the critical need for clinical research capacity in the Eastern Cape province. Situated strategically in East London's CBD, SBRI boasts a dedicated Research Team.
South African Health Products Regulatory Authority. As stated in the MRSA and ZAF-9, the South African Health Products Regulatory Authority (SAHPRA) is the regulatory authority overseeing medicines and clinical research, as well as medical devices and radiation safety. A s stated in the MRSA and GRMRSA, SAHPRA is responsible for clinical trial oversight, approval, and inspections in South Africa.
Select NIAID-Supported Research in South Africa: ... More than 2,500 participants enrolled at 34 clinical sites including South Africa, Kenya, Malawi, Uganda, and Zimbabwe. Shorter regimens enable patients to be cured faster and may reduce treatment costs, improve patient quality of life, increase therapy completion, and reduce development of ...
Clinresco Centres is a clinical research company that strives to provide service excellence to sponsoring pharmaceutical companies when conducting clinical research studies. We conduct clinical trials in South Africa according to approved protocols, International Conference on Harmonization-Good Clinical Practice (ICH-GCP), and South African-GCP guidelines.
The continent of Africa is home to 17% of the global population that carries 25% of the global disease burden. Yet globally, it accounts for less than 2% of genomic data1 used in medical innovation and less than 3% of clinical trials.2 Clinical research remains concentrated in high-income countries.3 Here, we highlight the importance of diversity in clinical research and propose solutions to ...
As leaders in clinical trial management, WHC assists with clinical research activities, site logistics, support, document storage and overall trial management. We provide all necessary support for client negotiations onsite to assess the feasibility of trials and to secure contract. Once approved, we can allocate study co-ordinators, clinical ...
Get more information about our trials from one of our friendly, qualified medical professionals and see how we can help you! Contact Us. 7th Floor, The Views, Tygervalley Health Centre, 43 Old Oak Rd, Tygervalley, 7530. 021 957 9400. 079 578 4731.
Below are actively recruiting clinical trials for South Africa. Click on the closest city to find the research studies that are available in your area. 000775 Durbanville. 1 total. 007925 Wynberg. 1 total. 173 Rivonia Road, Sandton. 1 total. 323 Umhlanga Rocks Drive, Suite 7, 3rd Floor, Umhlanga.
South African Clinical Research Association, Johannesburg. 402 likes · 2 talking about this. SACRA is a non-profit organisation representing the clinical research industry in South Africa
The facility, Nzimande added, will enhance South Africa's research in medicinal chemistry and this will expedite the development of drugs to address national priority diseases such as cancer and TB.
DHCT has developed and manages the HIV Clinicians Expert Telemedicine Platform, which is the largest HIV telemedicine platform in South Africa, covering the entire country (and not just Cape Town ...
Also, she has mentored students and trainees in North America and South Africa in rheumatology research. She is a USA representative on the Paediatric Musculoskeletal Task Force. She has published widely on lupus in Africa and the challenges to rheumatology clinical care, education, and practice in less-resourced countries.