Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Perspective
- Published: 23 November 2021

The probabilistic model of Alzheimer disease: the amyloid hypothesis revised
- Giovanni B. Frisoni ORCID: orcid.org/0000-0001-7075-7082 1 , 2 ,
- Daniele Altomare ORCID: orcid.org/0000-0003-1905-8993 1 , 2 ,
- Dietmar Rudolf Thal ORCID: orcid.org/0000-0002-1036-1075 3 , 4 ,
- Federica Ribaldi ORCID: orcid.org/0000-0001-9208-4472 1 , 2 , 5 , 6 ,
- Rik van der Kant 7 , 8 ,
- Rik Ossenkoppele 7 , 9 ,
- Kaj Blennow ORCID: orcid.org/0000-0002-1890-4193 10 ,
- Jeffrey Cummings 11 ,
- Cornelia van Duijn ORCID: orcid.org/0000-0002-2374-9204 12 , 13 ,
- Peter M. Nilsson ORCID: orcid.org/0000-0002-5652-8459 14 ,
- Pierre-Yves Dietrich 15 ,
- Philip Scheltens 7 , 16 &
- Bruno Dubois 17 , 18
Nature Reviews Neuroscience volume 23 , pages 53–66 ( 2022 ) Cite this article
21k Accesses
182 Citations
109 Altmetric
Metrics details
- Alzheimer's disease
- Molecular neuroscience
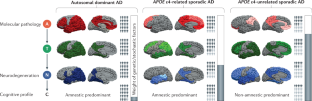
The current conceptualization of Alzheimer disease (AD) is driven by the amyloid hypothesis, in which a deterministic chain of events leads from amyloid deposition and then tau deposition to neurodegeneration and progressive cognitive impairment. This model fits autosomal dominant AD but is less applicable to sporadic AD. Owing to emerging information regarding the complex biology of AD and the challenges of developing amyloid-targeting drugs, the amyloid hypothesis needs to be reconsidered. Here we propose a probabilistic model of AD in which three variants of AD (autosomal dominant AD, APOE ε4-related sporadic AD and APOE ε4-unrelated sporadic AD) feature decreasing penetrance and decreasing weight of the amyloid pathophysiological cascade, and increasing weight of stochastic factors (environmental exposures and lower-risk genes). Together, these variants account for a large share of the neuropathological and clinical variability observed in people with AD. The implementation of this model in research might lead to a better understanding of disease pathophysiology, a revision of the current clinical taxonomy and accelerated development of strategies to prevent and treat AD.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
176,64 € per year
only 14,72 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Identification of senescent, TREM2-expressing microglia in aging and Alzheimer’s disease model mouse brain

APOE4/4 is linked to damaging lipid droplets in Alzheimer’s disease microglia

Molecular and cellular mechanisms of selective vulnerability in neurodegenerative diseases
Prince, M. et al. World Alzheimer Report 2015. The Global Impact of Dementia - An Analysis of Prevalence, Incidence, Cost and Trends . https://www.alzint.org/u/WorldAlzheimerReport2015.pdf (2015).
Ballard, C. et al. Alzheimer’s disease. Lancet 377 , 1019–1031 (2011).
PubMed Google Scholar
Jack, C. R. et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14 , 535–562 (2018).
PubMed PubMed Central Google Scholar
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256 , 184–185 (1992).
CAS PubMed Google Scholar
Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8 , 595–608 (2016).
CAS PubMed PubMed Central Google Scholar
Sorel, N., Cayssials, É., Brizard, F. & Chomel, J. C. Treatment and molecular monitoring update in chronic myeloid leukemia management. Ann. Biol. Clin. 75 , 129–145 (2017).
Google Scholar
Trojanowski, J. Q. Tauists, baptists, syners, apostates, and new data. Ann. Neurol. 52 , 263–265 (2002).
Makin, S. The amyloid hypothesis on trial. Nature 559 , S4–S7 (2018).
De Strooper, B. & Karran, E. The cellular phase of Alzheimer’s disease. Cell 164 , 603–615 (2016).
Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18 , 794–799 (2015).
Glenner, G. G. & Wong, C. W. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 122 , 1131–1135 (1984).
Bateman, R. J. et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Res. Ther. 3 , 1 (2011).
Jonsson, T. et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488 , 96 (2012).
Liu, C. C., Kanekiyo, T., Xu, H. & Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9 , 106–118 (2013).
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18 , 421–430 (2016).
Sleegers, K. & Van Duijn, C. M. Alzheimer’s disease: genes, pathogenesis and risk prediction. Community Genet. 4 , 197–203 (2001).
Ringman, J. et al. Neuropathology of autosomal dominant Alzheimer disease in the National Alzheimer Coordinating Center database. J. Neuropathol. Exp. Neurol. 75 , 284–290 (2016).
Jack, C. R. et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9 , 119–128 (2010).
van der Kant, R., Goldstein, L. S. B. & Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21 , 21–35 (2020).
Götz, J., Chen, F., Van Dorpe, J. & Nitsch, R. M. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 293 , 1491–1495 (2001).
Lewis, J. et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293 , 1487–1491 (2001).
Gomes, L. A. et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 138 , 913–941 (2019).
Choi, S. H. et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515 , 274–278 (2014).
Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H. & LaFerla, F. M. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43 , 321–332 (2004).
Jack, C. R. et al. The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 142 , 3230–3242 (2019).
La Joie, R. et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 12 , eaau5732 (2020).
Bancher, C. et al. Accumulation of abnormally phosphorylated τ precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 477 , 90–99 (1989).
Koper, M. J. et al. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 139 , 463–484 (2020).
Wiersma, V. I. et al. Granulovacuolar degeneration bodies are neuron-selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol. 138 , 943–970 (2019).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25 , 486–541 (2018).
Tang, D. et al. The molecular machinery of regulated cell death. Cell Res. 29 , 347–364 (2019).
Hanseeuw, B. J. et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann. Neurol. 81 , 583–596 (2017).
Bejanin, A. et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140 , 3286–3300 (2017).
Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537 , 50–56 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02477800 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02484547 (2021).
Haeberlein, S. B. et al. EMERGE and ENGAGE topline results: two phase 3 studies to evaluate aducanumab in patients with early Alzheimer’s disease. https://investors.biogen.com/static-files/ddd45672-9c7e-4c99-8a06-3b557697c06f (2019).
ALZFORUM. Gantenerumab https://www.alzforum.org/therapeutics/gantenerumab (2021).
ALZFORUM. AADvac1 https://www.alzforum.org/therapeutics/aadvac1 (2021).
Villemagne, V. L. et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12 , 357–367 (2013).
Jack, C. R. et al. Brain β-amyloid load approaches a plateau. Neurology 80 , 890–896 (2013).
Swanson, C. J. et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 13 , 80 (2021).
Mintun, M. A. et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 384 , 1691–1704 (2021).
Cummings, J., Lee, G., Ritter, A., Sabbagh, M. & Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. Transl. Res. Clin. Interv. 5 , 272–293 (2019).
National Institute on Aging. NIA-funded active Alzheimer’s and related dementias clinical trials and studies. https://www.nia.nih.gov/research/ongoing-AD-trials#section2 (2021).
Altomare, D. et al. Applying the ATN scheme in a memory clinic population: the ABIDE project. Neurology 93 , E1635–E1646 (2019).
Soldan, A. et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology 92 , E1567–E1579 (2019).
Ebenau, J. L. et al. ATN classification and clinical progression in subjective cognitive decline. Neurology https://doi.org/10.1212/wnl.0000000000009724 (2020).
Article PubMed PubMed Central Google Scholar
Vos, S. J. B. et al. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 12 , 957–965 (2013).
Weigand, A. et al. Is tau in the absence of amyloid on the Alzheimer’s continuum?: a study of discordant PET positivity. Brain Commun. 2 , fcz046 (2020).
Rabinovici, G. D. et al. Distinct MRI atrophy patterns in autopsy-proven Alzheimer’s disease and frontotemporal lobar degeneration. Am. J. Alzheimers. Dis. Other Demen. 22 , 474–488 (2008).
Ekman, U., Ferreira, D. & Westman, E. The A/T/N biomarker scheme and patterns of brain atrophy assessed in mild cognitive impairment. Sci. Rep. 8 , 8431 (2018).
Adams, D., Koike, H., Slama, M. & Coelho, T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat. Rev. Neurol. 15 , 387–404 (2019).
Pascoal, T. A. et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain https://doi.org/10.1093/brain/awaa180 (2020).
Leuzy, A. et al. Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 77 , 955–965 (2020).
Crary, J. F. et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128 , 755–766 (2014).
Duyckaerts, C. et al. PART is part of Alzheimer disease. Acta Neuropathol. 129 , 749–756 (2015).
Braak, H., Thal, D. R., Ghebremedhin, E. & Del Tredici, K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70 , 960–969 (2011).
Spires-Jones, T. L., Attems, J. & Thal, D. R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 134 , 187–205 (2017).
Sturchler-Pierrat, C. et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl Acad. Sci. USA 94 , 13287–13292 (1997).
Games, D. et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373 , 523–527 (1995).
Hsiao, K. et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274 , 99–102 (1996).
Corbett, G. T. et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 139 , 503–526 (2020).
Salazar, S. V. et al. Conditional deletion of Prnp rescues behavioral and synaptic deficits after disease onset in transgenic Alzheimer’s disease. J. Neurosci. 37 , 9207–9221 (2017).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82 , 239–259 (1991).
Thal, D. R., Rüb, U., Orantes, M. & Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58 , 1791–1800 (2002).
Shi, Y. et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549 , 523–527 (2017).
Karanth, S. et al. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.1741 (2020).
Schneider, J. A., Arvanitakis, Z., Bang, W. & Bennett, D. A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69 , 2197–2204 (2007).
Imbimbo, B. P. & Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 28 , 967–975 (2019).
Hochhaus, A. et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 34 , 966–984 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01760005 (2021).
ALZFORUM. Solanezumab https://www.alzforum.org/therapeutics/solanezumab (2021).
Alzheimer’s Association. DIAN-TU phase 3 clinical trials, topline results–news https://www.alz.org/news/2020/dian-tu-phase-3-clinical-trials-topline-results (2020).
Eisai Co. Ltd. Eisai and Biogen announce presentation of additional data from the phase II clinical trial of BAN2401 in early Alzheimer’s disease and the 2018 Clinical Trials on Alzheimer’s Disease (CTAD) Conference https://www.eisai.com/news/2018/news201892.html (2018).
Murray, M. E. et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10 , 785–796 (2011).
Schott, J. M. et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement. 12 , 862–871 (2016).
Carrasquillo, M. M. et al. Late-onset Alzheimer disease genetic variants in posterior cortical atrophy and posterior AD. Neurology 82 , 1455–1462 (2014).
Miller, Z. A. et al. Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol. 75 , 728–737 (2018).
Miller, Z. A. et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 136 , 3461–3473 (2013).
Bateman, R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367 , 795–804 (2012).
Hanseeuw, B. J. et al. PET staging of amyloidosis using striatum. Alzheimers Dement. 14 , 1281–1292 (2018).
Stokin, G. B. et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s diseases. Science 307 , 1282–1288 (2005).
Johnson, V. E., Stewart, W. & Smith, D. H. Traumatic brain injury and amyloid-β pathology: a link to alzheimer’s disease? Nat. Rev. Neurosci. 11 , 361–370 (2010).
Ikonomovic, M. D. et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 190 , 192–203 (2004).
Roberts, G. W., Gentleman, S. M., Lynch, A. & Graham, D. I. βA4 amyloid protein deposition in brain after head trauma. Lancet 338 , 1422–1423 (1991).
Rossor, M. N., Fox, N. C., Mummery, C. J., Schott, J. M. & Warren, J. D. The diagnosis of young-onset dementia. Lancet Neurol. 9 , 793–806 (2010).
Ryman, D. C. et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 83 , 253–260 (2014).
Sanchez, J. S. et al. Longitudinal amyloid and tau accumulation in autosomal dominant Alzheimer’s disease: findings from the Colombia-Boston (COLBOS) biomarker study. Alzheimers Res. Ther. 13 , 27 (2021).
Gordon, B. A. et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 17 , 241–250 (2018).
Gordon, B. A. et al. Tau PET in autosomal dominant Alzheimer’s disease: relationship with cognition, dementia and other biomarkers. Brain 142 , 1063–1076 (2019).
Cash, D. M. et al. The pattern of atrophy in familial Alzheimer disease: volumetric MRI results from the DIAN study. Neurology 81 , 1425–1433 (2013).
Lloyd, G. M. et al. Prominent amyloid plaque pathology and cerebral amyloid angiopathy in APP V717I (London) carrier - phenotypic variability in autosomal dominant Alzheimer’s disease. Acta Neuropathol. Commun. 8 , 31 (2020).
Sutovsky, S. et al. Neuropathology and biochemistry of early onset familial Alzheimer’s disease caused by presenilin-1 missense mutation Thr116Asn. J. Neural Transm. 125 , 965–976 (2018).
Gondim, D. D. et al. Diffuse Lewy body disease and Alzheimer disease: neuropathologic phenotype associated with the PSEN1 p.A396T mutation. J. Neuropathol. Exp. Neurol. 78 , 585–594 (2019).
Lippa, C. F. et al. Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 153 , 1365–1370 (1998).
Mann, D. et al. Predominant deposition of amyloid-beta 42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am. J. Pathol. 148 , 1257–1266 (1996).
Mann, D. M. A. et al. Amyloid β protein (Aβ) deposition in chromosome 14-linked Alzheimer’s disease: predominance of Aβ(42(43)). Ann. Neurol. 40 , 149–156 (1996).
Taipa, R. et al. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: a post mortem study. Neuropathol. Appl. Neurobiol. 44 , 298–313 (2018).
Ryan, N. S. et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 15 , 1326–1335 (2016).
Arboleda-Velasquez, J. F. et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. 25 , 1680–1683 (2019).
Yu, C. E., Chen, S., Jayadev, S. & Bird, T. Lack of APOE Christchurch variant in five age of onset outliers with PSEN1, PSEN2 Alzheimer’s disease and MAPT frontotemporal dementia. J. Neurol. Sci. 418 , 117143 (2020).
Müller, S. et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer’s disease. Alzheimers Dement. 14 , 1427–1437 (2018).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15 , 501–518 (2019).
Mattsson, N. et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement. 14 , 913–924 (2018).
Myers, R. H. et al. Apolipoprotein E ε4 association with dementia in a population-based study: The Framingham Study. Neurology 46 , 673–677 (1996).
Slooter, A. J. C. et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch. Neurol. 55 , 964–968 (1998).
Belloy, M. E., Napolioni, V. & Greicius, M. D. A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101 , 820–838 (2019).
Corder, E. H. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261 , 921–923 (1993).
Roses, A. D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 47 , 387–400 (1996).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51 , 404–413 (2019).
Van Duijn, C. M. et al. Apolipoprotein E4 allele in a population–based study of early–onset Alzheimer’s disease. Nat. Genet. 7 , 74–78 (1994).
Collij, L. E. et al. Multitracer model for staging cortical amyloid deposition using PET imaging. Neurology 95 , e1538–e1553 (2020).
Ossenkoppele, R. et al. Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurology 80 , 359–365 (2013).
Lehmann, M. et al. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J. Neurol. Neurosurg. Psychiatry 85 , 266–273 (2014).
Burnham, S. C. et al. Impact of APOE-ε4 carriage on the onset and rates of neocortical Aβ-amyloid deposition. Neurobiol. Aging 95 , 46–55 (2020).
Toledo, J. B. et al. APOE effect on amyloid-β PET spatial distribution, deposition rate, and cut-points. J. Alzheimers Dis. 69 , 783–793 (2019).
Schmechel, D. E. et al. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl Acad. Sci. USA 90 , 9649–9653 (1993).
Olichney, J. M. et al. Relationship between severe amyloid angiopathy, apolipoprotein E genotype, and vascular lesions in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 903 , 138–143 (2000).
Thal, D. R. et al. Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 61 , 282–293 (2002).
Thal, D. R., Griffin, W. S. T., de Vos, R. A. I. & Ghebremedhin, E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathologica 115 , 599–609 (2008).
Thal, D. R. et al. Capillary cerebral amyloid angiopathy identifies a distinct APOE ε4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol. 120 , 169–183 (2010).
Greenberg, S. M. et al. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat. Rev. Neurol. 16 , 30–42 (2020).
Ossenkoppele, R. et al. Discriminative accuracy of [ 18 F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 320 , 1151–1162 (2018).
Frisoni, G. B. et al. The topography of grey matter involvement in early and late onset Alzheimer’s disease. Brain 130 , 720–730 (2007).
Mattsson, N. et al. Greater tau load and reduced cortical thickness in APOE ε4-negative Alzheimer’s disease: a cohort study. Alzheimers Res. Ther. 10 , 77 (2018).
Emrani, S., Arain, H. A., DeMarshall, C. & Nuriel, T. APOE4 is associated with cognitive and pathological heterogeneity in patients with Alzheimer’s disease: a systematic review. Alzheimers Res. Ther. 12 , 141 (2020).
Therriault, J. et al. Association of apolipoprotein E ε4 with medial temporal tau independent of amyloid-β. JAMA Neurol. 77 , 470–479 (2020).
Geroldi, C. et al. APOE-ε4 is associated with less frontal and more medial temporal lobe atrophy in AD. Neurology 53 , 1825–1832 (1999).
Nelson, P. T. et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142 , 1503–1527 (2019).
Twohig, D. et al. The relevance of cerebrospinal fluid α-synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol. Commun. 6 , 130 (2018).
Weintraub, S. et al. APOE is a correlate of phenotypic heterogeneity in Alzheimer disease in a national cohort. Neurology 94 , e607–e612 (2020).
Reiman, E. M. et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 11 , 667 (2020).
Genin, E. et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16 , 903–907 (2011).
Bellenguez, C. et al. New insights on the genetic etiology of Alzheimer’s and related dementia. medRxiv 17 , 10 (2020).
van der Lee, S. J. et al. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol. 17 , 434–444 (2018).
Desikan, R. S. et al. Genetic assessment of age-associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 14 , e1002258 (2017).
Chouraki, V. et al. Evaluation of a genetic risk score to improve risk prediction for Alzheimer’s disease. J. Alzheimers Dis. 53 , 921–932 (2016).
Holstege, H. et al. The 100-plus Study of cognitively healthy centenarians: rationale, design and cohort description. Eur. J. Epidemiol. 33 , 1229–1249 (2018).
Laurent, S., Boutouyrie, P., Cunha, P. G., Lacolley, P. & Nilsson, P. M. Concept of extremes in vascular aging: from early vascular aging to supernormal vascular aging. Hypertension 74 , 218–228 (2019).
Bruno, R. M. et al. Early and supernormal vascular aging. Hypertension 76 , 1616–1624 (2020).
Ding, Y.-N., Tang, X., Chen, H.-Z. & Liu, D.-P. Epigenetic regulation of vascular aging and age-related vascular diseases. Adv. Exp. Med. Biol. 1086 , 55–75 (2018).
Dang, C. et al. Relationship between amyloid-β positivity and progression to mild cognitive impairment or dementia over 8 years in cognitively normal older adults. J. Alzheimers Dis. 65 , 1313–1325 (2018).
Farrer, L. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 278 , 1349–1356 (1997).
Saddiki, H. et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: a cerebrospinal fluid biomarker–based case–control study. PLoS Med. 17 , e1003289 (2020).
Nelis, S. M. et al. The impact of co-morbidity on the quality of life of people with dementia: findings from the IDEAL study. Age Ageing 48 , 361–367 (2019).
Strittmatter, W. J. et al. Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 90 , 1977–1981 (1993).
Thal, D. R. et al. Occurrence and co-localization of amyloid β-protein and apolipoprotein E in perivascular drainage channels of wild-type and APP-transgenic mice. Neurobiol. Aging 28 , 1221–1230 (2007).
Deane, R. et al. apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118 , 4002–4013 (2008).
Damotte, V. et al. Plasma amyloid β levels are driven by genetic variants near APOE, BACE1, APP, PSEN2: a genome-wide association study in over 12,000 non-demented participants. Alzheimers Dement. https://doi.org/10.1002/alz.12333 (2021).
Wang, C. et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector article. Nat. Med. 24 , 647–657 (2018).
Davis, A. A. et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 12 , eaay3069 (2020).
Zhao, N. et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 12 , eaay1809 (2020).
Yang, H. S. et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE ε4 haplotype status: a community-based cohort study. Lancet Neurol. 17 , 773–781 (2018).
Montagne, A. et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 581 , 1–6 (2020).
Blanchard, J. W. et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 26 , 1–12 (2020).
Hecht, M., Krämer, L. M., von Arnim, C. A. F., Otto, M. & Thal, D. R. Capillary cerebral amyloid angiopathy in Alzheimer’s disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 135 , 681–694 (2018).
Dean, D. C. et al. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA Neurol. 71 , 11–22 (2014).
Reiman, E. M. et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the ε4 allele for apolipoprotein E. N. Engl. J. Med. 334 , 752–758 (1996).
Evans, S. L. et al. Mid age APOE ε4 carriers show memory-related functional differences and disrupted structure-function relationships in hippocampal regions. Sci. Rep. 10 , 3110 (2020).
Reiman, E. M. et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl Acad. Sci. USA 101 , 284–289 (2004).
Reale,, M. et al. Relationship between inflammatory mediators, Aβ levels and ApoE genotype in Alzheimer disease. Curr. Alzheimer Res. 9 , 447–457 (2012).
CAS Google Scholar
Gorelick, P. B. Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann. N. Y. Acad. Sci. 1207 , 155–162 (2010).
Morgan, A. R. et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 15 , 776–787 (2019).
Cattaneo, A. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49 , 60–68 (2017).
Heneka, M. T. et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14 , 388–405 (2015).
Friedberg, J. S. et al. Associations between brain inflammatory profiles and human neuropathology are altered based on apolipoprotein E ε4 genotype. Sci. Rep. 10 , 29624 (2020).
Sheng, J., Mrak, R. & Griffin, W. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J. Neuropathol. Exp. Neurol. 56 , 285–290 (1997).
Griffin, W. S. T., Sheng, J. G., Roberts, G. W. & Mrak, R. E. Interleukin-1 expression in different plaque types in Alzheimer’s disease significance in plaque evolution. J. Neuropathol. Exp. Neurol. 54 , 276–281 (1995).
Akiyama, H. et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21 , 383–421 (2000).
Scheltens, P. et al. Alzheimer’s disease. Lancet https://doi.org/10.1016/S0140-6736(20)32205-4 (2021).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63 , 168–174 (2006).
Livingston, G. et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396 , 413–446 (2020).
Sims, R., Hill, M. & Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 23 , 311–322 (2020).
Zhang, Q. et al. Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nat. Commun. 11 , 4799 (2020).
Andreone, B. J. et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat. Neurosci. 23 , 927–938 (2020).
Nugent, A. A. et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105 , 837–854.e9 (2020).
De Roeck, A., Van Broeckhoven, C. & Sleegers, K. The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 138 , 201–220 (2019).
Sims, R. et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49 , 1373–1384 (2017).
Wollmer, M. A. Cholesterol-related genes in Alzheimer’s disease. Biochim. Biophys. Acta 1801 , 762–773 (2010).
Cuyvers, E. & Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 15 , 857–868 (2016).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51 , 414–430 (2019).
van der Lee, S. J. et al. A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 138 , 237–250 (2019).
Magno, L. et al. Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res. Ther. 11 , 16 (2019).
Jiao, S. S. et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 6 , e907 (2016).
Belloy, M. E., Napolioni, V., Han, S. S., Le Guen, Y. & Greicius, M. D. Association of Klotho-VS heterozygosity with risk of Alzheimer disease in individuals who carry APOE4. JAMA Neurol. 77 , 849–862 (2020).
Satoh, J.-I. et al. TMEM106B expression is reduced in Alzheimer’s disease brains. Alzheimers. Res. Ther. 6 , 17 (2014).
Hohman, T. J., Koran, M. E. I. & Thornton-Wells, T. A. Genetic modification of the relationship between phosphorylated tau and neurodegeneration. Alzheimers Dement. 10 , 637–645.e1 (2014).
Licher, S. et al. Genetic predisposition, modifiable-risk-factor profile and long-term dementia risk in the general population. Nat. Med. 25 , 1364–1369 (2019).
Lourida, I. et al. Association of lifestyle and genetic risk with incidence of dementia. JAMA 322 , 430–437 (2019).
Solomon, A. et al. Effect of the apolipoprotein E genotype on cognitive change during a multidomain lifestyle intervention a subgroup analysis of a randomized clinical trial. JAMA Neurol. 75 , 462–470 (2018).
Bostanciklioğlu, M. The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 127 , 954–967 (2019).
Marizzoni, M., Provasi, S., Cattaneo, A. & Frisoni, G. B. Microbiota and neurodegenerative diseases. Curr. Opin. Neurol. 30 , 630–638 (2017).
Cryan, J. F., O’Riordan, K. J., Sandhu, K., Peterson, V. & Dinan, T. G. The gut microbiome in neurological disorders. Lancet Neurol. 19 , 179–194 (2020).
Arenaza-Urquijo, E. M. & Vemuri, P. Resistance vs resilience to Alzheimer disease. Neurology 90 , 695–703 (2018).
Dumitrescu, L. et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain 143 , 2561–2575 (2020).
Gladyshev, V. N. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15 , 594–602 (2016).
Partridge, L., Deelen, J. & Slagboom, P. E. Facing up to the global challenges of ageing. Nature 561 , 45–56 (2018).
Herndon, L. A. et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. Elegans. Nature 419 , 808–814 (2002).
Williams, G. C. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11 , 398–411 (1957).
Altmann, A., Tian, L., Henderson, V. W. & Greicius, M. D. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75 , 563–573 (2014).
Fisher, D. W., Bennett, D. A. & Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 70 , 308–324 (2018).
Shen, S., Zhou, W., Chen, X. & Zhang, J. Sex differences in the association of APOE ε4 genotype with longitudinal hippocampal atrophy in cognitively normal older people. Eur. J. Neurol. 26 , 1362–1369 (2019).
Morris, J. C. et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76 , 264–273 (2019).
Williams, T., Borchelt, D. R. & Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein e function in Alzheimer’s disease. Mol. Neurodegener. 15 , 8 (2020).
Xiong, M. et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 13 , eabd7522 (2021).
Langa, K. M. & Burke, J. F. Preclinical Alzheimer disease - early diagnosis or overdiagnosis? JAMA Intern. Med. 179 , 1161–1162 (2019).
Jicha, G. A. & Rentz, D. M. Cognitive and brain reserve and the diagnosis and treatment of preclinical Alzheimer disease. Neurology 80 , 1180–1181 (2013).
Frisoni, G. B. et al. Re-aligning scientific and lay narratives of Alzheimer’s disease. Lancet Neurol. 18 , 918–919 (2019).
Frisoni, G. B. et al. Precision prevention of Alzheimer’s and other dementias: anticipating future needs in the control of risk factors and implementation of disease-modifying therapies. Alzheimers Dement. https://doi.org/10.1002/alz.12132 (2020).
Tavana, J. P. et al. RAB10: an Alzheimer’s disease resilience locus and potential drug target. Clin. Interv. Aging 14 , 73–79 (2019).
Barroeta-Espar, I. et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 121 , 327–337 (2019).
Cummings, J., Lee, G., Ritter, A., Sabbagh, M. & Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. Transl. Res. Clin. Interv. 6 , e12050 (2020).
Ballard, C. et al. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 17 , 213–222 (2018).
Iqbal, K. & Grundke-Iqbal, I. Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J. Cell. Mol. Med. 12 , 38–55 (2008).
Veitch, D. P. et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 15 , 106–152 (2019).
Derry, P. J. et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 184 , 101716 (2020).
Download references
Acknowledgements
This Perspective was the result of a workshop funded by the Swiss National Science Foundation entitled “How many roads lead to Rome? Insights in Alzheimer disease pathophysiology to lead future drug development” (grant number IZSEZ0_192840). G.B.F. received funding from the following sources: European Prevention of Alzheimer’s Dementia - EPAD (grant agreement number 115736) and Amyloid Imaging to Prevent Alzheimer’s Disease - AMYPED (grant agreement number 115952) funded by the EU–EFPIA Innovative Medicines Initiatives 2 Joint Undertaking; the Swiss National Science Foundation (“Brain connectivity and metacognition in persons with subjective cognitive decline (COSCODE): correlation with clinical features and in vivo neuropathology” (grant number 320030_182772)); Association Suisse pour la Recherche sur la Maladie d’Alzheimer, Geneva; Fondation Segré, Geneva; I. Pictet, Geneva; Fondazione Agusta, Lugano; Fondation Chmielewski, Geneva; and the VELUX Foundation. D.R.T. received funding from Fonds Wetenschappelijk Onderzoek Vlaanderen (FWO-G0F8516N Odysseus). R.v.d.K. was supported by an Alzheimer Nederland pilot grant (WE.03-2017-08) and a grant from the Selfridges Group Foundation (NR170059). K.B. is supported by the Swedish Research Council (2017-00915), the Swedish Alzheimer Foundation (AF-742881), Hjärnfonden, Sweden (FO2017-0243), and the Swedish state under an agreement between the Swedish government and the county councils, the ALF agreement (ALFGBG-715986). J.C. is supported by Keep Memory Alive, NIGMS grant P20GM109025, NINDS grant U01NS093334 and NIA grant R01AG053798.
Author information
Authors and affiliations.
Laboratory of Neuroimaging of Aging (LANVIE), University of Geneva, Geneva, Switzerland
Giovanni B. Frisoni, Daniele Altomare & Federica Ribaldi
Memory Clinic, Geneva University Hospitals, Geneva, Switzerland
Laboratory for Neuropathology, Department of Imaging and Pathology, and Leuven Brain Institute, University of Leuven, Leuven, Belgium
Dietmar Rudolf Thal
Department of Pathology, University Hospital Leuven, Leuven, Belgium
Laboratory of Alzheimer’s Neuroimaging and Epidemiology (LANE), IRCCS Centro S. Giovanni di Dio Fatebenefratelli, Brescia, Italy
Federica Ribaldi
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy
Alzheimer Center Amsterdam, Department of Neurology, Amsterdam Neuroscience, Amsterdam UMC, Amsterdam, Netherlands
Rik van der Kant, Rik Ossenkoppele & Philip Scheltens
Center for Neurogenomics and Cognitive Research, Amsterdam Neuroscience, Vrije Universiteit Amsterdam, Amsterdam UMC, Amsterdam, Netherlands
Rik van der Kant
Clinical Memory Research Unit, Lund University, Lund, Sweden
Rik Ossenkoppele
Cinical Neurochemistry Laboratory, Institute of Neuroscience and Physiology, University of Gothenburg, Sahlgrenska University Hospital, Mölndal, Sweden
Kaj Blennow
Chambers-Grundy Center for Transformative Neuroscience, Department of Brain Health, School of Integrated Health Sciences; University of Nevada, Las Vegas, Las Vegas, NV, USA
Jeffrey Cummings
Department of Epidemiology, Erasmus University Medical Center, Rotterdam, Netherlands
Cornelia van Duijn
Clinical Trial Service Unit and Epidemiological Studies Unit, Nuffield Department of Population Health, University of Oxford, Oxford, UK
Department of Clinical Sciences, Lund University, Skåne University Hospital, Malmö, Sweden
Peter M. Nilsson
Department of Oncology, Geneva University Hospitals, Geneva, Switzerland
Pierre-Yves Dietrich
Life Science Partners, Amsterdam, Netherlands
Philip Scheltens
Institut de la Mémoire et de la Maladie d’Alzheimer, IM2A, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, Paris, France
Bruno Dubois
Institut du Cerveau et de la Moelle Épinière, UMR-S975, INSERM, Paris, France
You can also search for this author in PubMed Google Scholar
Contributions
The authors all researched data for the article, provided substantial contributions to discussion of its content and reviewed/edited the manuscript before submission. G.B.F., D.A., D.R.T., F.R., R.v.d.K., R.O., P.M.N., P.-Y.D., P.S. and B.D. wrote the article.
Corresponding author
Correspondence to Giovanni B. Frisoni .
Ethics declarations
Competing interests.
G.B.F. has received grants from Avid Radiopharmaceuticals, Biogen, GE International, Guerbert, IXICO, Merz Pharma, Nestlé, Novartis, Eisai, Piramal, Roche, Siemens, Teva Pharmaceutical Industries and Vifor Pharma. He has received personal fees from AstraZeneca, Avid Radiopharmaceuticals, Biogen, Roche, Diadem, Neurodiem, Elan Pharmaceuticals, GE International, Lundbeck, Pfizer and TauRx Therapeutics. D.R.T. has received speaker honoraria from Novartis Pharma Basel (Switzerland) and Biogen (USA), has received travel reimbursement from GE Healthcare (UK), and UCB (Belgium) and has collaborated with GE Healthcare (UK), Novartis Pharma Basel (Switzerland), Probiodrug (Germany) and Janssen Pharmaceuticals (Belgium). K.B. has served as a consultant, on advisory boards or on data monitoring committees for Abcam, Axon, Biogen, Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, which is part of the GU Ventures incubator programme. J.C. has acted as a consultant for Acadia, Actinogen, Alkahest, Alzheon, Annovis, Avanir, Axsome, Biogen, Cassava, Cerecin, Cerevel, Cortexyme, Cytox, EIP Pharma, Eisai, Foresight, GemVax, Genentech, Green Valley, Grifols, Karuna, Merck, Novo Nordisk, Otsuka, Resverlogix, Roche, Samumed, Samus, Signant Health, Suven and United Neuroscience. J.C. also has stock options in ADAMAS, AnnovisBio, MedAvante and BiOasis, and owns the copyright of the Neuropsychiatric Inventory. P.S. has received consultancy fees (paid to Amsterdam UMC) from AC Immune, Brainstorm Cell, EIP, ImmunoBrain Checkpoint, Genentech, Novartis, and Novo Noridisk. He is a principal investigator on studies with AC Immune, FUJIFILM Toyama, UCB, and Vivoryon. He is a part-time employee of Life Sciences Partners Amsterdam. B.D. has received research funding (paid to the institution) from Merck-Avenir Foundation and Roche and consultancy fees from Biogen, Neurodiem, Green Valley, Cytox and Brainstorm. He is a principal investigator on clinical trials with Eisai, Genentech, Novartis, Biogen and Roche. D.A., F.R., R.v.d.K., R.O., C.v.D., P.M.N. and P.-Y.D. declare no competing interests.
Additional information
Peer review information.
Nature Reviews Neuroscience thanks G. Bu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
(AD). The co-occurrence of brain Aβ and tau pathology. AD dementia is the final stage of AD, in which cognitive impairment and loss of daily function are also present.
In the brain, a 37–49-amino-acid polypeptide (amyloid-β (Aβ)) produced by the metabolism of the synaptic membrane protein amyloid precursor protein (APP). The amyloid fibrillar form is made mainly of the 42-amino-acid variant (Aβ 42 ) and is the primary component of amyloid plaques found in the brains of individuals with Alzheimer disease. Soluble Aβ 42 can be found in plasma and the cerebrospinal fluid and can give rise to soluble oligomers, thought to be the toxic form of Aβ.
Braak stage denotes the degree of tau pathology in Alzheimer disease and assumes progressive spread of such pathology from the transentorhinal region of the brain. Braak stages I and II denote neurofibrillary tangle involvement confined mainly to the transentorhinal region, stages III and IV when there is also involvement of limbic regions such as the hippocampus, and stages V and VI when there is extensive neocortical involvement.
(MCI). A syndrome featuring cognitive impairment and no loss of daily function; Alzheimer disease is the underlying pathology in 60–80% of MCI cases. In these cases, the condition is also called prodromal Alzheimer disease or MCI due to Alzheimer disease.
Progressive loss of the structure or function of neurons, which may ultimately involve cell death. The earliest detectable event is thought to be synaptic loss, followed by neuronal loss. Neurodegeneration can be detected in vivo with volumetric MRI and positron emission tomography with 18 F-labelled deoxyglucose.
A protein whose primary role is in maintaining the stability of microtubules in axons. In the course of Alzheimer disease, tau becomes hyperphosphorylated, leading to axonal and synaptic dysfunction and aggregation of tau into intracellular neurofibrillary tangles.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Frisoni, G.B., Altomare, D., Thal, D.R. et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci 23 , 53–66 (2022). https://doi.org/10.1038/s41583-021-00533-w
Download citation
Accepted : 04 October 2021
Published : 23 November 2021
Issue Date : January 2022
DOI : https://doi.org/10.1038/s41583-021-00533-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Neuropathological changes associated with aberrant cerebrospinal fluid p-tau181 and aβ42 in alzheimer’s disease and other neurodegenerative diseases.
- Masanori Kurihara
- Tomoyasu Matsubara
Acta Neuropathologica Communications (2024)
Data-driven modelling of neurodegenerative disease progression: thinking outside the black box
- Alexandra L. Young
- Neil P. Oxtoby
- Daniel C. Alexander
Nature Reviews Neuroscience (2024)
APOE3ch alleviates Aβ and tau pathology and neurodegeneration in the human APPNL-G-F cerebral organoid model of Alzheimer’s disease
Cell Research (2024)
Epilepsy and epileptiform activity in late-onset Alzheimer disease: clinical and pathophysiological advances, gaps and conundrums
- Anita Kamondi
- Madeleine Grigg-Damberger
- Andras Attila Horvath
Nature Reviews Neurology (2024)
Association of APOE gene with longitudinal changes of CSF amyloid beta and tau levels in Alzheimer’s disease: racial differences
- Danqing Xiao
- Kesheng Wang
Neurological Sciences (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

- Open access
- Published: 30 January 2018
Alzheimer’s disease hypothesis and related therapies
- Xiaoguang Du 1 ,
- Xinyi Wang 1 &
- Meiyu Geng 2
Translational Neurodegeneration volume 7 , Article number: 2 ( 2018 ) Cite this article
23k Accesses
378 Citations
2 Altmetric
Metrics details
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common cause for dementia. There are many hypotheses about AD, including abnormal deposit of amyloid β (Aβ) protein in the extracellular spaces of neurons, formation of twisted fibers of tau proteins inside neurons, cholinergic neuron damage, inflammation, oxidative stress, etc., and many anti-AD drugs based on these hypotheses have been developed. In this review, we will discuss the existing and emerging hypothesis and related therapies.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, which is the most common cause for dementia and imposes immense suffering on patients and their families. According to the World Alzheimer Report 2016, there are currently about 46.8 million people suffering with AD worldwide. The ageing of world population will further compound this problem and lead to a steep increase in the number of AD patients. The numbers of AD patients are expected to double nearly every 20 years, and thereby the population of AD will reach 74.7 million in 2030 and 131.5 million in 2050 [ 1 ]. AD has become the third major cause of disability and death for the elderly, only after cardiovascular and cerebrovascular diseases and malignant tumors.
However, only five drugs have been approved by the FDA to treat AD over the past hundred years since the first AD patient was diagnosed. Not only that, these approved drugs including cholinesterase inhibitors, N-methyl-D-aspartate (NMDA) receptor antagonist or their combination usually provide temporary and incomplete symptomatic relief accompanied with severe side effects. The marginal benefits were unable to slow the progression of AD. Thus, developing drugs for more effective AD treatment is in urgent need.
Current hypothesis about AD and anti-AD drug development
AD is a complicated disease involving many factors. Due to the complexity of human brains, the lack of reasonable animal models and research tools, the detailed pathogenesis of AD is still unclear so far. Many hypotheses about AD have been developed, including amyloid β (Aβ), Tau, cholinergic neuron damage and oxidative stress, inflammation, etc. Thus, many efforts have been done to develop anti-AD drugs based on these hypotheses.
Aβ cascade hypothesis
Extracellular deposits of Aβ peptides as senile plaques, intraneuronal neurofibrillary tangles (NFTs), and large-scale neuronal loss were the main pathological features of AD. Thus, Aβ peptides have long been viewed as a potential target for AD which dominated new drug research during the past twenty years [ 2 ]. The most direct strategy in anti-Aβ therapy is to reduce Aβ production by targeting β- and γ-secretase [ 3 ]. Safety issues are the overriding problem. For targeting γ-secretase, undesirable side effects are inevitable due to its physiological substrates, eg. the Notch signaling protein [ 4 , 5 , 6 , 7 ], which is essential in normal biological process. Similarily, targeting β-secretase is also challenged for the side effects such as blindness and the large catalytic pocket [ 8 ]. More importantly, in sporadic AD cases, the majority of AD patients do not necessarily have over-producted amyloid precursor protein. Besides, Aβ isoforms could also serve as endogenous positive regulators for neurotransmitter release at hippocampal synapses [ 9 ]. Thus, inhibiting Aβ production may encounter many challenges.
Aβ clearance by immunotherapy is the alternative choice. For active Aβ-immunotherapy, although the first active AD vaccine (AN1792) developed by ELAN showed some beneficial effects such as less cognitive decline, it was suspended owing to serious side effect, or meningoencephalitis [ 10 , 11 , 12 ]. Also, the passive immunotherapy did not do much better than active immunotherapy. Several antibodies targeting Aβ have failed in clinical trials, including bapineuzumab (Pfizer/Johnson & Johnson) [ 13 , 14 ], Crenezumab (Genentech) [ 15 , 16 ], solanezumab (Eli Lilly) [ 16 , 17 , 18 ] and ponezumab (Johnson & Johnson /Pfizer) [ 19 , 20 , 21 ]. In addition, although passive immunotherapy could overcome some problems of active immunotherapy, there were still inevitable side effects such as amyloid-related imaging abnormalities [ 22 ]. Likewise, the small molecule Aβ binder scyllo-inositol [ 23 ] and tramiprosate [ 24 , 25 , 26 ] also failed in clinical trials. These failures even cast more doubts on the Aβ theory [ 27 ]. Actually, the strategy of targeting only a single functional subregion of Aβ may partly account for these failures [ 27 , 28 ]. Furthermore, immunotherapy may influence the human immune system, which might cause beneficial or detrimental consequence (such as side effects). However, every cloud has a silver lining. A phase Ib trial of aducanumab (Biogen) showed a positive correlation between brain Aβ levels and disease exacerbation as measured by Clinical Dementia Rating [ 29 , 30 , 31 ]. Even the failed phase III EXPEDITION3 trial of solanezumab (Eli Lilly) still demonstrated better performance in Clinical Dementia Rating Sum of Boxes and beneficial impacts on Mini-Mental State Examination and Activities of Daily Living [ 17 , 18 , 32 , 33 ]. Thus, despite all kinds of problems, immunotherapy may still be the better approach to modify the extent of neurodegeneration in AD currently [ 34 ].
In fact, the original amyloid cascade hypothesis was that “Aβ is the causative agent in Alzheimer’s Disease pathology, and that neurofibrillary tangles, cell loss, vascular damage, and dementia follow as a direct result of this deposition” [ 35 ]. After decades of research, although the bulk of data still supports a role for Aβ as the primary initiator of the complex pathogenic cascade in AD, more and more evidences indicate that Aβ acts as a trigger in the early disease process and appears to be necessary but not sufficient in the late stage of AD [ 36 ]. Especially, recent rapid progresses in understanding on toxic amyloid assembly and Aβ metabolism associated systemic abnormalities will provide fresh impetus and new opportunities for this interesting approach [ 37 ].
Tau hypothesis
Neurofibrillary tangles, another intracellular hallmark of AD, are composed of tau. Tau is a microtubule-associated protein working as scaffolding proteins that are enriched in axons. In pathological conditions, tau aggregation will impair axons of neurons and thus cause neurodegeneration. After numerous failures of Aβ-targeting drugs for AD, more interests are turning to explore the therapeutic potential of targeting tau, particularly as studies of biomarkers suggest that tau pathology is more closely linked to the progression of AD [ 38 ].
Tau undergoes many modifications, including phosphorylation, arginine monomethylation, lysine acetylation, lysine monomethylation, lysine dimethylation, lysine ubiquitylation and serine.
O-linked N-acetylglucosamine (O-GlcNAc) modification [ 39 ]. Under pathological conditions, increasing of tau hyperphosphorylation will render the protein aggregation-proned, reduce its affinity for microtubules, and thereby influence neuronal plasticity. Consequently, strategies to target tau involve blocking of tau aggregation, utilizing tau vaccinations, stabilizing microtubules, manipulating kinases and phosphatases that govern tau modifications. However, most of these efforts have failed in clinical trials. For Tau aggregation blockers, TRx0237 failed to show treatment benefits in phase III trials [ 40 ]. As for vaccinations, tau-targeted active vaccines (ACI35 and AADvac-1) and passive vaccines (RG6100 and ABBv-8E12) are currently in phase I and II clinical trials [ 41 , 42 ]. Intravenous immunoglobulin (IVIG), the only passive vaccine in phase III clinical trials, failed to meet the primary end points in patients with mild-to-moderate AD [ 42 ]. Other tau-targeting strategies for AD, including stabilizing microtubules and manipulating kinases and phosphatases, have just been tested in preclinical studies.
In general, tau-targeting therapies remain challenging because of incomplete understanding of AD, lack of robust and sensitive biomarkers for diagnosis and response-monitoring, and the obstruction of blood-brain barrier.
Inflammation hypothesis
Reactive gliosis and neuroinflammation are hallmarks of AD. Microglia-related pathways were considered to be central to AD risk and pathogenesis, as supported by emerging genetic and transcriptomic studies [ 43 , 44 , 45 , 46 , 47 ]. Increasing evidence demonstrate that microglia emerges as central players in AD. In very early stage, microglia, TREM2 and complement system are responsible for synaptic pruning [ 48 , 49 ]. The processes of activity-dependent and long-term synaptic plasticity are the common and fundamental cellular underpinning of learning and memory which may manifest as influence on long term potential [ 50 ]. Following that, reactive microglia and astrocytes will surround amyloid plaques and secrete numerous pro-inflammatory cytokines. These events are regarded as an early, prime mover in AD evolution. However, non-steroid anti-inflammatory drugs (NSAIDs) did not show enough benefits in clinic. This is because that the relationship between innate immunity and AD pathogenesis is complex, and the immune response can be either deleterious or beneficial depending on the context [ 47 , 51 , 52 ]. However, the new observations that PD-1 immune checkpoint blockade reduces the pathology of AD and improves memory in mouse models of AD [ 53 , 54 , 55 ] give us a direction of future researches.
The recent advances in our understanding of the mechanism underlying microglia dysfunction in pruning, regulating plasticity, and neurogenesis are opening up possibilities for new opportunities of AD therapeutic interventions and diagnosis [ 56 , 57 ]. Targeting these aberrant microglial functions and thereby returning homeostasis may yield novel paradigms for AD therapies. However, given the complexity and diverse functions of microglia in health and disease, there is a crucial need for new biomarkers reflecting the function of specific microglias [ 52 , 58 ].
Cholinergic and oxidative stress hypothesis
Acetylcholine (ACh) is an important neurotransmitter used by cholinergic neurons, which has been involved in critical physiological processes, such as attention, learning, memory, stress response, wakefulness and sleep, and sensory information [ 59 , 60 , 61 , 62 , 63 ]. Cholinergic neurons damage was considered to be a critical pathological change that correlated with cognitive impairment in AD. Thus, cholinergic hypothesis was firstly tested with cholinesterase inhibitors in AD treatment. Tacrine, a cholinesterase inhibitor, was the first anti-AD drug available in clinic [ 64 , 65 , 66 ] although it was withdrawn from the market in 2012 due to severe side effects. Although inhibiting cholinesterase is a symptomatic relief treatment with marginal benefits, it is currently the most available clinical treatment which gives desperate AD patients a glimmer of hope. For other neurotransmitter dysfunction, such as Dopamine and 5-hydroxytryptamine, there are some studies about them, but not much as acetylcholine in AD.
Oxidative stress is considered to play an important role in the pathogenesis of AD. Especially, the brain utilizes more oxygen than other tissues and undergoes mitochondrial respiration, which increases the potential for ROS exposure. In fact, AD is highly associated with cellular oxidative stress, including augmentation of protein oxidation, protein nitration, glycoloxidation and lipid peroxidation as well as accumulation of Aβ, for Aβ can also induce oxidative stress [ 67 , 68 , 69 , 70 , 71 , 72 , 73 ]. Thus, the treatment with anti-oxidant compounds would provide protection against oxidative stress and Aβ toxicity in theory. However, oxidative stress is only a single feature of AD, so antioxidant strategy was challenged for its potency to stop the progression of AD and thus it is proposed as a portion of combination therapy [ 74 , 75 ].
Glucose hypometabolism
Glucose hypometabolism is the early pathogenic event in the prodromal phase of AD, and associated with cognitive and functional decline. Early therapeutic intervention before the irreversible degeneration has become a consensus in AD treatment. Thus, alleviation of glucose hypometabolism was emerged as an attractive strategy of AD treatment. However, most of these therapeutic strategies are targeting mitochondria and bioenergetics, which have shown promise at the preclinical stage but without success in clinical trials [ 76 , 77 ]. Although no strategies are available to alleviate glucose hypometabolism in clinical, glucose metabolism brain imaging such as 18 FDG-PET (Positron emision tomography with 2-deoxy-2-fluorine-18-fluoro-D-glucose) has become a valuable indicator for diagnosis of neurodegenerative diseases that cause dementia, including AD [ 78 ].
Up to now, there’re no effective treatments for changing the course of AD. Confronting these difficulties, we should get deeper understandings about these hypotheses, and meanwhile we should renovate our knowledge about AD and develop new hypothesis.
New pathway to AD
AD is conventionally regarded as a central nervous system (CNS) disorder. However, increasing experimental, epidemiological and clinical evidences have suggested that manifestations of AD extend beyond the brain. Most notably, research over the past few years reveals that the gut microbiome (GMB) has a profound impact on the formation of the blood-brain barrier, myelination, neurogenesis, and microglia maturation [ 79 , 80 , 81 , 82 , 83 , 84 ]. In particular, results from germ-free animals and animals exposed to pathogenic microbial infections, antibiotics, probiotics, or fecal microbiota transplantation showed that gut microbiota modulates many aspects of animal behaviors, suggesting a role for the gut microbiota in host cognition or AD-related pathogenesis [ 85 , 86 , 87 , 88 ]. The underlying mechanisms of gut microbiota influencing brain involve the communication through immune system, the endocrine system, the vague nerve, and the bacteria-derived metabolites.
Immune pathway
The intestinal mucosal lymphoid tissue contains 70% ~ 80% of the immune cells in the whole body, and is considered to be the largest and most important human immune organs. It is also the first line of host defense against pathogens. The human gut contains a large, diverse and dynamic enteric microbiota, including more than 100 trillion microorganisms from at least 1000 distinct species. There’s a complex relationship between intestinal mucosal immune system and intestinal microbiota. Thus, gut microbiota induced immunomodulation is emerging as an important pathway that influences AD [ 89 ].
Gut microbiota can influence brain through immune system in several ways. Firstly, intestinal microbiome can induce cytokines secretion, which enter the circulatory system, pass through blood brain barrier, and directly affect the brain function. For instance, perivascular macrophages and cerebral small vessel epithelial cells can receive the intestinal microbiome produced IL-1 signal and affect central nervous system. Also, gut microbes can activate Toll-like receptors of the brain immune cells (such as microglia) through microbes associated molecular patterns (MAMP). MAMPs can either directly bind to intestinal epithelial cells or infiltrate to the intestine lamina propria to activate lymphocytes, promoting the release of pro-inflammatory cytokines, which further cause subsequent inflammation in brain. Secondly, gut microbes can produce metabolites such as short-chain-fatty acids (SCFAs), gamma-aminobutyric acid (GABA) and 5-HT precursors, which could also travel to the brain via circulatory systems or signal through intestinal epithelials to produce cytokines or neurotransmitters that activate vagus nerve. Thirdly, gut microbes can activate enteroendocrine cells to produce 5-HT, which affect the brain through neuroimmune pathways.
In addition to changing the functions of the immune system, such as through secretion of inflammatory factors or anti-inflammatory factors, intestinal microbiome can also affect the development and composition of immune system. For example, in germ-free mice, isolated lymphoid follicles in gut associated lymphoid tissue are unable to mature, and lymphocytes that are able to secrete IgA in the intestinal epithelium decreased [ 89 , 90 , 91 , 92 ]. For immune system in brain, the deletion of gut microbiota in germ-free mice have global influence on the cell proportions and maturation of microglia in the brain, and thus affect the properties and phenotype of microglia, as compared to conventionally colonized controls [ 93 ]. Similar results were obtained in antibiotic treated mice. Other research also demonstrates that the number of T regulatory cells and T helper lymphocytes (T helper 17, Th17) are significantly reduced in the germ free mouse, indicating the regulatory effects of intestinal microbiome on T cell composition, while microbiome tansplant to germ free mice can modify these variations and restore normal immune function [ 94 , 95 ]. All these modulations of gut microbiota may have direct and indirect effects on AD development and progression.
Endocrine pathway and the vagus nerve
The gut is also the largest endocrine organ in the body. Gut microbiota can regulate secretion of many hormones from intestinal endocrine cells, such as corticosterone and adrenal hormones, and thus establish the information exchange between the intestines and the brain. For example, the intestinal microbiome can affect the secretion of serotonin and regulate brain emotional activities [ 96 , 97 ]; intestinal microbial metabolism can also produce a variety of neurotransmitters, such as dopamine, GABA, acetylcholine and melatonin, which are transmitted to central nervous system through the vagus nerve [ 98 ]. Besides transporting these signal substances, the vagus nerve itself plays an important role in inflammation and depression [ 99 ]. The vagus nerve can influence the gastrointestinal tract, orchestrate the complex interactions between central and peripheral neural control mechanisms [ 100 ]. The stimulation of vagus nerve is able to regulate mood, and the immune system, suggesting the therapeutic potential of vagus nerve modulation to attenuate the pathophysiological changes and restore homeostasis [ 98 , 99 , 100 , 101 , 102 , 103 ].
Bacteria-derived metabolites
Generation of essential nutrients for host physiology, such as vitamins and other cofactors, is an important physiological function of the gut microbiota [ 104 ]. The metabolites of microbiome, such as SCFAs including acetate, butyrate, and propionate, are able to modulate peripheral and central pathologic processes [ 105 ]. For example, butyrate is effective in reducing inflammation and pain. Once in the brain, acetate is able to alter the level of the neurotransmitters glutamate, glutamine, and GABA, as well as increases anorectic neuropeptide expression [ 106 ]. In addition, the gut microbiota can secrete large amounts of amyloids and lipopolysaccharides, which might contribute to the modulation of signaling pathways, the production of proinflammatory cytokines associated with AD pathogenesis and Aβ deposition [ 107 , 108 , 109 ].
In fact, microbiota-gut-brain axis has been established and a disturbed gut microbiota has been incriminated in many neurodegenerative diseases in animal and translational models. In theory, a role for the microbiota-gut-brain axis is highly plausible. However, the theoretical basis for the use of microbiota-directed therapies in neurodegenerative disorders still needs supports from high-quality clinical trials [ 110 ]. To date, only a few studies directly focused on the gut microbiota and AD [ 111 , 112 ], and studies on AD patients is particullarly deficient. A recent research from human showed an increase in the abundance of a pro-inflammatory GMB taxon and a reduction in the abundance of an anti-inflammatory taxon are possibly associated with a peripheral inflammatory state in patients with cognitive impairment and brain amyloidosis. It is important for the research of gut microbiota and AD. However, further investigations are still necessary to explore the possible causal relation between GMB-related inflammation and amyloidosis [ 111 ]. The comprehensive understanding of these underlying mechanisms may provide new insights into these novel therapeutic strategies for AD. In particular, based on the gut microbiota hypothesis, Chinese traditional medicine and probiotic bacteria may play a more important role in therapy [ 113 ].
Conclusions
Nowadays, new technologies are making it possible to get to know enough pathologic details of disease. More importantly, scientists are beginning to treat AD as a systemic disease and they are paying more attention to the correlation between brain and other organs [ 47 , 89 , 114 ]. Perhaps, for complicated disease such as AD, researches and therapies should be based on the principle that combined reductionism with holism, and great efforts should be made to search the fundamental laws of AD by means of multi-scale modeling and efficient numeric assessment. Maybe, just like Chinese traditional medicine [ 115 ], combination treatments or systematic therapy will be a final way out.
Abbreviations
Alzheimer’s disease
Central nervous system
Colony stimulating factor 1 receptor
Gamma-aminobutyric acid
Intravenous immunoglobulin
Microbes associated molecular patterns
Neurofibrillary tangles
N-methyl-D-aspartate
Non-steroid anti-inflammatory drugs
O-linked N-acetylglucosamine
Short-chain-fatty acids
T helper lymphocytes 17
Ali G-C, Guerchet M, Wu Y-T, Prince M, Prina M. Chapter 2: The global prevalence of dementia. In: Prince M, Guerchet M, Ali G-C, Wu Y-T, Prina M, editors. The Global Impact of Dementia. An analysis of prevalence, incidence, cost and trends. London: Alzheimer’s Disease International (ADI); 2015. p. 10–29.
Cummings J, et al. Drug development in Alzheimer’s disease: the path to 2025. Alzheimers Res Ther. 2016;8:39.
Article PubMed PubMed Central Google Scholar
Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27(3):419–22.
Article CAS PubMed Google Scholar
van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959–63.
Olry A, et al. Generation and characterization of mutant cell lines defective in gamma-secretase processing of notch and amyloid precursor protein. J Biol Chem. 2005;280(31):28564–71.
Tarassishin L, et al. Processing of notch and amyloid precursor protein by gamma-secretase is spatially distinct. Proc Natl Acad Sci U S A. 2004;101(49):17050–5.
Article CAS PubMed PubMed Central Google Scholar
Sastre M, et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of notch. EMBO Rep. 2001;2(9):835–41.
Klaver DW, et al. Is BACE1 a suitable therapeutic target for the treatment of Alzheimer’s disease? Current strategies and future directions. Biol Chem. 2010;391(8):849–59.
Abramov E, et al. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12(12):1567–76.
Gilman S, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in AN interrupted trial. Neurology. 2005;64(9):1553–62.
Bayer AJ, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64(1):94–101.
Holmes C, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–23.
Laskowitz DT, Kolls BJ. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2010;74(24):2026. author reply 2026-7
Article PubMed Google Scholar
Salloway S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–70.
Ultsch M, et al. Structure of Crenezumab complex with Abeta shows loss of beta-hairpin. Sci Rep. 2016;6:39374.
Bouter Y, et al. Abeta targets of the biosimilar antibodies of Bapineuzumab, Crenezumab, Solanezumab in comparison to an antibody against Ntruncated Abeta in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol. 2015;130(5):713–29.
The Lancet N. Solanezumab: too late in mild Alzheimer’ss disease? Lancet Neurol. 2017;16(2):97.
Article Google Scholar
Gandy S, Sano M. Alzheimer disease: Solanezumab-prospects for meaningful interventions in AD? Nat Rev Neurol. 2015;11(12):669–70.
Landen JW, et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: a phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin Neuropharmacol. 2013;36(1):14–23.
Burstein AH, et al. Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol. 2013;36(1):8–13.
La Porte SL, et al. Structural basis of C-terminal beta-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer's disease. J Mol Biol. 2012;421(4–5):525–36.
Carlson C, et al. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer's disease. Alzheimers Dement (Amst). 2016;2:75–85.
Google Scholar
Salloway S, et al. A phase 2 randomized trial of ELND005, scyllo-inositol, in mild to moderate Alzheimer disease. Neurology. 2011;77(13):1253–62.
Aisen PS, et al. Tramiprosate in mild-to-moderate Alzheimer's disease - a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase study). Arch Med Sci. 2011;7(1):102–11.
Greenberg SM, et al. A phase 2 study of tramiprosate for cerebral amyloid angiopathy. Alzheimer Dis Assoc Disord. 2006;20(4):269–74.
Gervais F, et al. Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28(4):537–47.
Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712.
Nie Q, Du XG, Geng MY. Small molecule inhibitors of amyloid beta peptide aggregation as a potential therapeutic strategy for Alzheimer's disease. Acta Pharmacol Sin. 2011;32(5):545–51.
Sevigny J, et al. Addendum: the antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2017;546(7659):564.
Sevigny J, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537(7618):50–6.
Patel KR. Biogen's aducanumab raises hope that Alzheimer's can be treated at its source. Manag Care. 2015;24(6):19.
Karran E. Recent trial shows that solanezumab has disease modifying effects. BMJ. 2015;351:h4528.
Article PubMed CAS Google Scholar
Siemers ER, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement. 2016;12(2):110–20.
Barrera-Ocampo A, Lopera F. Amyloid-beta immunotherapy: the hope for Alzheimer disease? Colomb Med (Cali). 2016;47(4):203–12.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5.
Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci. 2015;18(6):800–6.
Wang J, et al. A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612–23.
Brier MR, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. 2016;8(338):338ra66.
Article PubMed PubMed Central CAS Google Scholar
Morris M, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci. 2015;18(8):1183–9.
Gauthier S, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer's disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388(10062):2873–84.
Novak P, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer's disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16(2):123–34.
Li C, Gotz J. Tau-based therapies in neurodegeneration: opportunities and challenges. Nat Rev Drug Discov. 2017;
Zhang B, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153(3):707–20.
Guerreiro R, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368(2):117–27.
Song W, et al. Alzheimer's disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017;13(4):381–7.
Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. 2016;17(4):201–7.
Bolos M, Perea JR, Avila J. Alzheimer's disease as an inflammatory disease. Biomol Concepts. 2017;8(1):37–43.
Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–34.
Paolicelli RC, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–8.
Bliss TV, Collingridge GL, Morris RG. Synaptic plasticity in health and disease: introduction and overview. Philos Trans R Soc Lond Ser B Biol Sci. 2014;369(1633):20130129.
Article CAS Google Scholar
Chen J, et al. Inhibition of AGEs/RAGE/rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-kappaB pathway. J Neuroimmunol. 2017;305:108–14.
Hirbec H E, Noristani HN, Perrin FE. Microglia Responses in Acute and Chronic Neurological Diseases: What Microglia-Specific Transcriptomic Studies Taught (and did Not Teach) Us. Front Aging Neurosci. 2017;9:227.
Baruch K, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med. 2016;22(2):135–7.
Saresella M, et al. A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer's disease. Neurobiol Aging. 2012;33(3):624 e11–22.
Saresella M, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;21(3):927–38.
Jevtic S, et al. The role of the immune system in Alzheimer disease: etiology and treatment. Ageing Res Rev. 2017;40:84–94.
McGeer PL, McGeer EG. Targeting microglia for the treatment of Alzheimer's disease. Expert Opin Ther Targets. 2015;19(4):497–506.
Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23(9):1018–27.
Hasselmo ME, Anderson BP, Bower JM. Cholinergic modulation of cortical associative memory function. J Neurophysiol. 1992;67(5):1230–46.
Fine A, et al. Learning impairments following injection of a selective cholinergic immunotoxin, ME20.4 IgG-saporin, into the basal nucleus of Meynert in monkeys. Neuroscience. 1997;81(2):331–43.
Sarter M, Bruno JP. Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Res Brain Res Rev. 1997;23(1–2):28–46.
Miranda MI, Bermudez-Rattoni F. Reversible inactivation of the nucleus basalis magnocellularis induces disruption of cortical acetylcholine release and acquisition, but not retrieval, of aversive memories. Proc Natl Acad Sci U S A. 1999;96(11):6478–82.
Haam J, Yakel JL. Cholinergic modulation of the hippocampal region and memory function. J Neurochem. 2017;142(Suppl 2):111–21.
Brinkman SD, Gershon S. Measurement of cholinergic drug effects on memory in Alzheimer's disease. Neurobiol Aging. 1983;4(2):139–45.
Summers WK, et al. Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type. N Engl J Med. 1986;315(20):1241–5.
Summers WK, et al. Use of THA in treatment of Alzheimer-like dementia: pilot study in twelve patients. Biol Psychiatry. 1981;16(2):145–53.
CAS PubMed Google Scholar
Cheignon C, et al. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450–64.
Sultana R, Butterfield DA. Redox proteomics studies of in vivo amyloid beta-peptide animal models of Alzheimer's disease: insight into the role of oxidative stress. Proteomics Clin Appl. 2008;2(5):685–96.
Butterfield DA, et al. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43(5):658–77.
Mohmmad Abdul H, et al. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1-42), HO and kainic acid: implications for Alzheimer's disease. J Neurochem. 2006;96(5):1322–35.
Gibson GL, Allsop D, Austen BM. Induction of cellular oxidative stress by the beta-amyloid peptide involved in Alzheimer's disease. Protein Pept Lett. 2004;11(3):257–70.
Butterfield DA, et al. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer's disease brain exists. J Alzheimers Dis. 2002;4(3):193–201.
Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32(11):1050–60.
Persson T, Popescu BO, Cedazo-Minguez A. Oxidative stress in Alzheimer's disease: why did antioxidant therapy fail? Oxidative Med Cell Longev. 2014;2014:427318.
Teixeira J, et al. Alzheimer's disease and antioxidant therapy: how long how far? Curr Med Chem. 2013;20(24):2939–52.
Caldwell CC, Yao J, Brinton RD. Targeting the prodromal stage of Alzheimer's disease: bioenergetic and mitochondrial opportunities. Neurotherapeutics. 2015;12(1):66–80.
Daulatzai MA. Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer's disease. J Neurosci Res. 2017;95(4):943–72.
Nasrallah I, Dubroff J. An overview of PET neuroimaging. Semin Nucl Med. 2013;43(6):449–61.
Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64(1):61–78.
Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89.
Lui H, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–35.
Elmer BM, McAllister AK. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci. 2012;35(11):660–70.
Erickson MA, Dohi K, Banks WA. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation. 2012;19(2):121–30.
Banks WA. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav Immun. 2015;44:1–8.
Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol. 2012;10(11):735–42.
Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701–12.
Sampson TR, Mazmanian SK. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 2015;17(5):565–76.
Bhattacharjee S, Lukiw WJ. Alzheimer's disease and the microbiome. Front Cell Neurosci. 2013;7:153.
Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. 2017;46(4):562–76.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–41.
Tomkovich S, Jobin C. Microbiota and host immune responses: a love-hate relationship. Immunology. 2016;147(1):1–10.
Agace WW, McCoy KD. Regionalized development and maintenance of the intestinal adaptive immune landscape. Immunity. 2017;46(4):532–48.
Erny D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–77.
Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98.
Sudo N, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol. 2004;558(Pt 1):263–75.
O'Mahony SM, et al. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. 2015;277:32–48.
Praveen V, Praveen S. Microbiome-gut-brain Axis: a pathway for improving brainstem serotonin homeostasis and successful autoresuscitation in SIDS-A novel hypothesis. Front Pediatr. 2016;4:136.
PubMed Google Scholar
Jiang XG, et al. Research progress in anti-inflammation of vagus nerve and neurotransmitter ach. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2003;15(1):59–61.
Das UN. Vagus nerve stimulation, depression, and inflammation. Neuropsychopharmacology. 2007;32(9):2053–4.
Browning KN, Verheijden S, Boeckxstaens GE. The Vagus nerve in appetite regulation, mood, and intestinal inflammation. Gastroenterology. 2017;152(4):730–44.
de Haan JJ, et al. Exploring the link between inflammation and the vagus nerve. J Intern Med. 2010;267(1):130–1.
Hoeger S, et al. Modulation of brain dead induced inflammation by vagus nerve stimulation. Am J Transplant. 2010;10(3):477–89.
Mirakaj V, et al. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J Exp Med. 2014;211(6):1037–48.
Gordon JI, et al. The human gut microbiota and undernutrition. Sci Transl Med. 2012;4(137):137ps12.
Mitchell RW, et al. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem. 2011;117(4):735–46.
Frost G, et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun. 2014;5:3611.
Zhao Y, Dua P, Lukiw WJ. Microbial sources of amyloid and relevance to Amyloidogenesis and Alzheimer's disease (AD). J Alzheimers Dis Parkinsonism. 2015;5(1):177.
CAS PubMed PubMed Central Google Scholar
Schwartz K, Boles BR. Microbial amyloids--functions and interactions within the host. Curr Opin Microbiol. 2013;16(1):93–9.
Hill JM, Lukiw WJ. Microbial-generated amyloids and Alzheimer's disease (AD). Front Aging Neurosci. 2015;7:9.
Quigley EMM. Microbiota-brain-gut Axis and neurodegenerative diseases. Curr Neurol Neurosci Rep. 2017;17(12):94.
Cattaneo A, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–8.
Park AM, et al. Helicobacter pylori and gut microbiota in multiple sclerosis versus Alzheimer's disease: 10 pitfalls of microbiome studies. Clin Exp Neuroimmunol. 2017;8(3):215–32.
Chen F, et al. Could the gut microbiota reconcile the oral bioavailability conundrum of traditional herbs? J Ethnopharmacol. 2016;179:253–64.
Zheng X, et al. Thinking outside the brain for cognitive improvement: Is peripheral immunomodulation on the way? Neuropharmacology. 2015;96(Pt A):94–104.
Yang WT, et al. Chinese herbal medicine for Alzheimer's disease: clinical evidence and possible mechanism of neurogenesis. Biochem Pharmacol. 2017;141:143–55.
Download references
Acknowledgements
Not applicable.
This review was supported by “Personalized Medicines-Molecular Signature based Drug Discovery and Development”, Strategic Priority Research program of Sciences, Grants No. XDA12040101.
Availability of data and materials
Author information, authors and affiliations.
Shanghai GreenValley Pharmaceutical Co., Ltd., 421 Newton Road, Shanghai, 201203, People’s Republic of China
Xiaoguang Du & Xinyi Wang
State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai, 201203, People’s Republic of China
You can also search for this author in PubMed Google Scholar
Contributions
All authors read and approved the final manuscript. XD: Forming the concept, drafting and revising the manuscript; XW: Revising the immune pathway; MG: Revising and approving the manuscript.
Corresponding author
Correspondence to Meiyu Geng .
Ethics declarations
Ethics approval and consent to participate.
Not relevant.
Consent for publication
Competing interests.
The authors declare they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/ ), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated.
Reprints and permissions
About this article
Cite this article.
Du, X., Wang, X. & Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener 7 , 2 (2018). https://doi.org/10.1186/s40035-018-0107-y
Download citation
Received : 07 November 2017
Accepted : 18 January 2018
Published : 30 January 2018
DOI : https://doi.org/10.1186/s40035-018-0107-y
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Alzheimer’s disease - complicated disease - anti-AD drugs
- Hypothesis about AD
Translational Neurodegeneration
ISSN: 2047-9158
- Submission enquiries: Access here and click Contact Us
- General enquiries: [email protected]
Advertisement
25 Years of the Amyloid Hypothesis of the Origin of Alzheimer’s Disease: Advances, Failures, and New Perspectives
- Published: 17 November 2017
- Volume 47 , pages 1065–1070, ( 2017 )
Cite this article

- O. S. Levin 1 &
- E. E. Vasenina 1
191 Accesses
3 Citations
Explore all metrics
The amyloid hypothesis of the development of Alzheimer’s disease (AD) continues to dominate, though the concept has changed significantly during its 25-year history. The accumulation of β-amyloid has been found to be linked not only with increase in its production (as found after elucidation of the genetic mechanisms of some familial cases of AD), but also with impairments to its clearance from brain tissues, which is mediated by the microcirculatory system. The most significant pathogenetic role in brain substance is played not by the senile plaques themselves, described by Alois Alzheimer almost 110 years ago and consisting of insoluble conjugates, but by soluble β-amyloid oligomers. The relationship between the vascular and degenerative processes in AD is supported by the common risk factors and by clinical, neuroimaging, pathomorphological, and experimental data. One component linking degenerative and vascular processes in AD is insulin resistance. Challenges of new multimodal therapeutic strategies for AD are discussed in relation to the current status of the amyloid hypothesis.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes

Is Alzheimer disease a disease?
Β-amyloid peptides and amyloid plaques in alzheimer’s disease.
D. Selkoe, “Resolving controversies on the path to Alzheimer’s therapeutics,” Nat. Med. , 17 , No. 9, 1060–1065 (2011); doi: https://doi.org/10.1038/nm.2460 .
Article CAS PubMed Google Scholar
G. Glenner and C. Wong, “Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein,” Biochem. Biophys. Res. Com. , 120 , No. 3, 885–890 (1984); doi: https://doi.org/10.1016/s0006-291x(84)80190-4 .
J. Hardy and D. Allsop, “Amyloid deposition as the central event in the aetiology of Alzheimer’s disease,” Trends Pharmacol. Sci. , 12 383–388 (1991); doi: https://doi.org/10.1016/0165-6147(91)90609-v .
D. Selkoe and J. Hardy, “The amyloid hypothesis of Alzheimer’s disease at 25 years,” EMBO Mol. Med. , 8 , No. 6, 595–608 (2016); doi: 10.15252/emmm.201606210 .
Article CAS PubMed PubMed Central Google Scholar
M. Ikram, “Risk prediction of Alzheimer’s disease: An epidemiologic perspective,” Alz. Dement. , 6 , No. 4, 119–120 (2010); doi: https://doi.org/10.1016/j.jalz.2010.05.373 .
Article Google Scholar
J. Luchsinger, C. Reitz, L. Honig, et al., “Aggregation of vascular risk factors and risk of incident Alzheimer’s disease,” Neurology , 65 , No. 4, 545–551 (2005); doi: https://doi.org/10.1212/01.wnl.0000172914.08967.dc .
S. Vermeer, N. Prins, T. den Heijer, et al., “Silent brain infarcts and the risk of dementia and cognitive decline,” N. Engl. J. Med. , 348 , No. 13, 1215–1222 (2003); doi: https://doi.org/10.1056/nejmoa022066 .
Article PubMed Google Scholar
B. Reed, S. Villeneuve, W. Mack, et al., “Associations between serum cholesterol levels and cerebral amyloidosis,” JAMA Neurol. , 71 , No. 2, 195 (2014); doi: https://doi.org/10.1001/jamaneurol.2013.5390 .
Article PubMed PubMed Central Google Scholar
K. Rodrigue, J. Rieck, K. Kennedy, et al., “Risk factors for β-amyloid deposition in healthy aging,” JAMA Neurol. , 70 , No. 5, 600 (2013); doi: https://doi.org/10.1001/jamaneurol.2013.1342 .
D. Barnes and K. Yaffe, “The projected effect of risk factor reduction on Alzheimer’s disease prevalence,” Lancet Neurol. , 10 , No. 9, 819–828 (2011); doi: https://doi.org/10.1016/S1474-4422(11)70072-2 .
D. Snowdon, “Brain infarction and the clinical expression of Alzheimer’s disease. The Nun Study,” JAMA , 277 , No. 10, 813–817 (1997); doi: https://doi.org/10.1001/jama.277.10.813 .
L. Honig, W. Kukull, and R. Mayeux, “Atherosclerosis and AD: Analysis of data from the US National Alzheimer’s Coordinating Center,” Neurology , 64 , No. 3, 494–500 (2005); doi: https://doi.org/10.1212/01.wnl.0000150886.50187.30 .
H. Dolan, B. Crain, J. Troncoso, et al., “Atherosclerosis, dementia, and Alzheimer’s disease in the BLSA cohort,” Ann. Neurol. , 68 , No. 2, 231–240 (2010); doi: https://doi.org/10.1002/ana.22055 .
PubMed PubMed Central Google Scholar
L. Zheng, H. Vinters, W. Mack, et al., “Cerebral atherosclerosis is associated with cystic infarcts and microinfarcts but not Alzheimer pathologic changes,” Stroke , 44 , No. 10, 2835–2841 (2013); doi: https://doi.org/10.1161/STROKEAHA.113.001945 .
H. Chui, L. Zheng, B. Reed, et al., “Vascular risk factors and Alzheimer’s disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review,” Alzheimer’s Res. Ther. , 3 , No. 6, 36 (2011); doi: https://doi.org/10.1186/alzrt98 .
J. Toledo, S. Arnold, K. Raible, et al., “Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre,” Brain , 136 , No. 9, 2697–2706 (2013); doi: https://doi.org/10.1093/brain/awt188 .
A. Roher, “Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease,” Arterioscler. Thromb. Vasc. Biol. , 23 , No. 11, 2055–2062 (2003); doi: https://doi.org/10.1161/01.atv.0000095973.42032.44 .
T. Beach, J. Wilson, L. Sue, et al., “Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles,” Acta Neuropathol. , 113 , No. 1, 13–21 (2006); doi: https://doi.org/10.1007/s00401-006-0136-y .
M. Yarchoan, S. Xie, M. Kling, et al., “Cerebrovascular atherosclerosis correlates with Alzheimer’s pathology in neurodegenerative dementias,” Brain , 135 , No. 12, 3749–3756 (2012); doi: https://doi.org/10.1093/brain/aws271 .
J. Neltner, E. Abner, S. Baker, et al., “Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing,” Brain , 137 , No. 1, 255–267 (2013); doi: https://doi.org/10.1093/brain/awt318 .
T. Thomas, S. Miners, and S. Love, “Post-mortem assessment of hypoperfusion of cerebral cortex in Alzheimer’s disease and vascular dementia,” Brain , 138 , No. 4, 1059–1069 (2015); doi: https://doi.org/10.1093/brain/awv025 .
A. Roher, Z. Garami, S. Tyas, et al., “Transcranial Doppler ultrasound blood flow velocity and pulsatility index as systemic indicators for Alzheimer’s disease,” Alz. Dement. , 7 , No. 4, 445–455 (2011); doi: https://doi.org/10.1016/j.jalz.2010.09.002 .
T. Hughes, L. Kuller, E. Barinas-Mitchell, et al., “Arterial stiffness and β-amyloid progression in nondemented elderly adults,” JAMA Neurol. , 71 , No. 5, 562 (2014).
X. Zhang, K. Zhou, R. Wang, et al., “Hypoxia-inducible factor 1 (HIF-1)-mediated hypoxia increases BACE1 expression and beta-amyloid generation,” J. Biol. Chem. , 282 , No. 15, 10,873–10,880 (2007); doi: https://doi.org/10.1074/jbc.m608856200 .
Article CAS Google Scholar
G. Tesco, Y. Koh, E. Kang, et al., “Depletion of GGA3 stabilizes BACE and enhances β-secretase activity,” Neuron , 54 , No. 5, 721–737 (2007); doi: https://doi.org/10.1016/j.neuron.2007. 05.012.
L. Li, X. Zhang, D. Yang, et al., “Hypoxia increases Aβ generation by altering β- and γ-cleavage of APP,” Neurobiol. Aging , 30 , No. 7, 1091–1098 (2009); doi: https://doi.org/10.1016/j.neurobiolaging.2007.10.011 .
Y. Okamoto, T. Yamamoto, R. Kalaria, et al., “Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts,” Acta Neuropathol. , 123 , No. 3, 381–394 (2011); doi: https://doi.org/10.1007/s00401-011-0925-9 .
H. Zetterberg, E. Mortberg, L. Song, et al., “Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans,” PLoS ONE , 6, No. 12, 28263 (2011); doi: https://doi.org/10.1371/journal.pone.0028263 .
G. Faraco, L. Park, P. Zhou, et al., “Hypertension enhances Aβ-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP,” J. Cereb. Blood Flow Metab. , 36 , No. 1, 241–252 (2015); doi: https://doi.org/10.1038/jcbfm.2015.79 .
C. Iadecola, “Vascular and metabolic factors in Alzheimer’s disease and related dementias,” Cell. Mol. Neurobiol. , 36 , 151–154 (2016); doi: https://doi.org/10.1007/s10571-015-0319-y.
K. Niwa, K. Kazama, L. Younkin, et al., “Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein,” Am. J. Physiol. Heart Circ. Physiol. , 283 , No. 1, 315–323 (2002); doi: https://doi.org/10.1152/ajpheart.00022.2002 .
T. Kuninaka, Y. Senga, H. Senga, and M. Weiner, “Nature of enhanced mitochondrial oxidative metabolism by a calf blood extract,” J. Cell. Physiol. , 146 , No. 1, 148–155 (1991); doi: https://doi.org/10.1002/jcp.1041.460119 .
S. Hoyer and K. Betz, “Elimination of the delayed postischemic energy deficit in cerebral cortex and hippocampus of aged rats with a dried, deproteinized blood extract (Actovegin),” Arch. Gerontol. Geriatr. , 9 , No. 2, 181–192 (1989); doi: https://doi.org/10.1016/0167-4943(89)90038-1 .
S. Jacob, G. J. Dietze, F. Machicao, et al., “Improvement of glucose metabolism in patients with type II diabetes after treatment with hemodialysate. Arzneimittelforschung , 3 , 269–272 (1996).
Google Scholar
M. Elmlinger, M. Kriebel, and D. Ziegler, “Neuroprotective and anti-oxidative effects of the hemodialysate Actovegin on primary rat neurons in vitro.,” Neuromol. Med. , 13 , No. 4, 266–274 (2011); doi: https://doi.org/10.1007/s12017-011-8157-7 .
A. Dieckmann, M. Kriebel, E. Andriambeloson, et al., “Treatment with Actovegin improves sensory nerve function and pathology in streptozotocin-diabetic rats via mechanisms involving inhibition of PARP activation,” Exp. Clin. Endocrinol. Diabetes , 120 , No. 3, 132–138 (2011); doi: https://doi.org/10.1055/s-0031-1291248 .
PubMed Google Scholar
F. Machicao, D. F. Muresanu, H. Hundsberger, et al., “Pleiotropic neuroprotective and metabolic effects of Actovegin’s mode of action,” J Neurol. Sci. , 322 , No. 1, 222–227 (2012); doi: https://doi.org/10.1016/j.jns.2012.07.069 .
J. Donoghue, “Actovegin in the treatment of dementia: A systematic review and analysis of outcomes using the number needed to treat (NNT),” Clin. Pharmacol. Ther. , 22 , No. 4, 70–75 (2013).
A. Guekht, I. Skoog, A. Korczyn, et al., “A randomised, double-blind, placebo-controlled trial of Actovegin in patients with post-stroke cognitive impairment: ARTEMIDA study design,” Dement. Geriatr. Cogn. Disord. Extra. , 3 , No. 1, 459–467 (2013); doi: https://doi.org/10.1159/000357122 .
Download references
Author information
Authors and affiliations.
Russian Postgraduate Medical Academy, Moscow, Russia
O. S. Levin & E. E. Vasenina
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to O. S. Levin .
Additional information
Translated from Zhurnal Nevrologii i Psikhiatrii imeni S. S. Korsakova, Vol. 116, No. 6, Iss. II, Neurology and Psychiatry in the Elderly, pp. 3–9, June, 2016.
Rights and permissions
Reprints and permissions
About this article
Levin, O.S., Vasenina, E.E. 25 Years of the Amyloid Hypothesis of the Origin of Alzheimer’s Disease: Advances, Failures, and New Perspectives. Neurosci Behav Physi 47 , 1065–1070 (2017). https://doi.org/10.1007/s11055-017-0513-0
Download citation
Published : 17 November 2017
Issue Date : November 2017
DOI : https://doi.org/10.1007/s11055-017-0513-0
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Alzheimer’s disease
- arteriosclerosis
- insulin resistance
- multimodal therapy
- Find a journal
- Publish with us
- Track your research
Alzheimer's disease: as it was in the beginning
Affiliation.
- PMID: 28704198
- DOI: 10.1515/revneuro-2017-0006
Since Alzheimer's disease was first described in 1907, many attempts have been made to reveal its main cause. Nowadays, two forms of the disease are known, and while the hereditary form of the disease is clearly caused by mutations in one of several genes, the etiology of the sporadic form remains a mystery. Both forms share similar sets of neuropathological and molecular manifestations, including extracellular deposition of amyloid-beta, intracellular accumulation of hyperphosphorylated tau protein, disturbances in both the structure and functions of mitochondria, oxidative stress, metal ion metabolism disorders, impairment of N-methyl-D-aspartate receptor-related signaling pathways, abnormalities of lipid metabolism, and aberrant cell cycle reentry in some neurons. Such a diversity of symptoms led to proposition of various hypotheses for explaining the development of Alzheimer's disease, the amyloid hypothesis, which postulates the key role of amyloid-beta in Alzheimer's disease development, being the most prominent. However, this hypothesis does not fully explain all of the molecular abnormalities and is therefore heavily criticized. In this review, we propose a hypothetical model of Alzheimer's disease progression, assuming a key role of age-related mitochondrial dysfunction, as was postulated in the mitochondrial cascade hypothesis. Our model explains the connections between all the symptoms of Alzheimer's disease, with particular attention to autophagy, metal metabolism disorders, and aberrant cell cycle re-entry in neurons. Progression of the Alzheimer's disease appears to be a complex process involving aging and too many protective mechanisms affecting one another, thereby leading to even greater deleterious effects.
Keywords: amyloid beta; autophagy; iron; mitochondrial dysfunction; neurofibrillary tangles; neuronal cell cycle re-entry; β-amyloid.
Publication types
- Alzheimer Disease / etiology*
- Alzheimer Disease / genetics
- Alzheimer Disease / metabolism
- Alzheimer Disease / pathology
- Amyloid beta-Peptides / genetics
- Amyloid beta-Peptides / metabolism
- Metals / metabolism
- Mitochondria / metabolism
- tau Proteins / genetics
- tau Proteins / metabolism
- Amyloid beta-Peptides
- tau Proteins
IMAGES
VIDEO
COMMENTS
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause for dementia. There are many hypotheses about AD, including abnormal deposit of amyloid β (Aβ) protein in the extracellular spaces of neurons, formation of twisted fibers of tau proteins inside neurons, cholinergic neuron damage, inflammation ...
2.1. The Amyloid-Beta Hypothesis. This hypothesis is the most recognized one amongst researchers, owing to its explanation for the senile plaque formation and the accumulation of Aβ oligomers as the major highlight of the disease [].The proteolysis of transmembrane protein APP by beta and gamma secretases forms single units of Aβ, which further undergo certain structural modifications to ...
Beta-amyloid and the amyloid hypothesis. In Alzheimer's disease, brain cells that process, store and retrieve information degenerate and die. Although scientists do not yet know the underlying cause of this destruction, they have identified several possible culprits. One prime suspect is a microscopic brain protein fragment called beta ...
Alzheimer's disease (AD) is a neurodegenerative disease characterized by progressive memory loss along with neuropsychiatric symptoms and a decline in activities of daily life. Its main ...
The amyloid hypothesis (AH) is still the most accepted model to explain the pathogenesis of inherited Alzheimer's disease (IAD). However, despite the neuropathological overlapping with the non-inherited form (NIAD), AH waver in explaining NIAD. Thus, 30 years after its first statement several questions are still open, mainly regarding the role ...
In the pathogenesis of Alzheimer's disease, the Aβ cascade hypothesis is the most popular theory. One of the key pathogenic features of AD is the accumulation of Aβ to create amyloid plaque ...
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause for dementia. There are many hypotheses about AD, including abnormal deposit of amyloid β (Aβ) protein in the extracellular spaces of neurons, formation of twisted fibers of tau proteins inside neurons, cholinergic neuron damage, inflammation ...
Alzheimer's disease is the most common form of dementia and the most common neurodegenerative disease. It manifests as a decline in short-term memory and cognition that impairs daily behavior. Most cases of Alzheimer's disease are sporadic, but a small minority of inherited forms allow gene identification which, together with neuropathology, yields important clues about the wider causes ...
The amyloid cascade hypothesis of Alzheimer disease (AD), which proposes that deposition of the amyloid-β (Aβ) peptide in the brain is a central event in disease pathology (Fig. 1), is strongly ...
The amyloid cascade hypothesis of Alzheimer disease (AD), which proposes that deposition of the amyloid-β (Aβ) peptide in the brain is a central event in disease pathology (Fig. 1), is strongly ...
The pathogenesis of Alzheimer's disease (AD) is currently unclear and is the subject of much debate. The most widely accepted hypothesis designed to explain AD pathogenesis is the amyloid cascade, which invokes the accumulation of extracellular plaques and intracellular tangles as playing a fundamental role in the course and progression of the disease.
Alzheimer disease (AD) is the most common cause of dementia in elderly people and is becoming increasingly prevalent worldwide. The incidence of dementia doubles with every six years of age, from ...
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause for dementia. There are many hypotheses about AD, including abnormal deposit of amyloid β (Aβ) protein in the extracellular spaces of neurons, formation of twisted fibers of tau proteins inside neurons, cholinergic neuron damage, inflammation, oxidative stress, etc., and many anti-AD drugs based ...
The amyloid hypothesis of the development of Alzheimer's disease (AD) continues to dominate, though the concept has changed significantly during its 25-year history. The accumulation of β-amyloid has been found to be linked not only with increase in its production (as found after elucidation of the genetic mechanisms of some familial cases of AD), but also with impairments to its clearance ...
Over the past 2 decades, the so-called amyloid hypothesis dominated the field of Alzheimer disease (AD) research. This theory states that AD pathology starts by the sequential cleavage of APP (amyloid precursor protein), which results in Aβ (amyloid β) accumulation, as plaques in brain parenchyma or as vascular deposits leading to cerebral amyloid angiopathy (CAA). 1 An imbalance in Aβ ...
The Science of Alzheimer's Disease Explained. Aug 2, 2022. Alzheimer's is a chronic, progressive neurodegenerative disease that affects at least 50 million people worldwide. The number of people living with Alzheimer's has increased dramatically since 1990 due to aging and population growth and is projected to exceed 152 million cases by ...
1. Introduction. Alzheimer's disease (AD) is the most prominent type of dementia. This neurodegenerative disorder affects an estimated 50 million individuals worldwide, and this figure is expected to triple by 2050 [].The current annual financial cost of AD care in the United States alone is estimated to be $300 billion [].Furthermore, many family members of affected patients are forced to ...
The amyloid hypothesis has so far been at the forefront of explaining the pathogenesis of Alzheimer's Disease (AD), a progressive neurodegenerative disorder that leads to cognitive decline and eventual death. Recent evidence, however, points to additional factors that contribute to the pathogenesis …
The 2-hit hypothesis of AD. The cell-cycle hypothesis of AD proposes a 2-hit hypothesis that results in neuron "immortality", and continual production of senile plaques and neurofibrillary tangles to cause AD. Neurons are able to leave G0 quiescence and enter a permanent, steady-state G1 phase. Neurons lose the ability to undergo apoptosis.
The vascular hypothesis of Alzheimer's disease (VHAD) was proposed 24 years ago from observations made in our laboratory using aging rats subjected to chronic brain hypoperfusion. In recent years, VHAD has become a mother-lode to numerous neuroimaging studies targeting cerebral hemodynamic changes, particularly brain hypoperfusion in elderly ...
2.1.1. Autosomal Dominant AD: The genetics of autosomal dominant AD is well-described with three predominant genes identified. Presenilin 1 (PSEN1) accounts for about 80% of autosomal dominant cases.Presenilin 2 (PSEN2) is quite rare, except in families of Volga German ancestry, accounting for only 5% of cases.About 15% of autosomal dominant cases are due to mutations in the amyloid precursor ...
Old age increases the risk of Alzheimer's disease (AD), the most common neurodegenerative disease, a devastating disorder of the human mind and the leading cause of dementia. Worldwide, 50 million people have the disease, and it is estimated that there will be 150 million by 2050. Today, healthcare for AD patients consumes 1% of the global economy. According to the amyloid cascade hypothesis ...
Abstract. Since Alzheimer's disease was first described in 1907, many attempts have been made to reveal its main cause. Nowadays, two forms of the disease are known, and while the hereditary form of the disease is clearly caused by mutations in one of several genes, the etiology of the sporadic form remains a mystery.