- Lupe_Grau Cancel

Laboratory Products for “Research Use Only” (RUO) – Often a Dangerous Claim
Manufactures use the “Research Use Only” (RUO) label to declare that their products should not be used in diagnostic procedures. This enables them to avoid the time-consuming and costly documentation required for conformity-assessed in vitro diagnostic medical devices (CE-IVDs). Nevertheless, some medical laboratories, for example, still use RUO products in diagnostic procedures, sometimes even with the knowledge of the manufacturers. This can have consequences – not just for manufacturers and operators, but for patients as well.
In this article, you will learn:
- What the “Research Use Only” label (RUO) means
- What the requirements for RUO products are
- How to avoid legal problems
- What alternatives there are to RUO products
1. “Research Use Only” – what does it mean?
Labeling products for “research use only” has far-reaching consequences. It means the products are barely subject to any regulatory controls under the IVDR. As a result, for a lot of manufacturers and operators, they are desirable alternatives to more costly and time-intensive conformity-assessed in-vitro diagnostic medical devices (CE-IVDs) that must comply with the applicable legal requirements.
a) Institutions affected
The following institutions, in particular, use RUO products:
- Medical laboratories can use RUO products, but this makes them the manufacturer with all the consequences this entails. You can find more information on “lab developed tests” in our article “ The E U Regulates Medical Laboratories. Are Laboratory Developed Tests Still Allowed? ”
- If medical laboratories use RUO products for purposes other than research then, in the worst case, this makes them liable for damages as well as criminally liable.
- Therefore, medical laboratories should inform themselves about the parameters for RUO products and possible alternatives .
- Manufacturers Manufacturers use RUO products as components for their IVDs. They should, therefore, make sure that they know all the requirements in detail before labeling a product as “RUO”.
b) Definition
There is no uniform definition of “research use only” products. In general, they can be understood to be what the name implies, i.e., products to be used for analysis that are intended to be used for scientific research purposes only.
They primarily differ from medical devices in that they cannot be used for medical purposes.
However, the understanding of “research use only” is different in Europe and the USA.

Definition in Europe
In Europe, the MEDDEV 2.14/2 guidance document (IVD Guidance: Research Use Only products – A guide for manufacturers and notified bodies) provides clues as to the definition of RUOs. This guidance was written within the framework of the now obsolete Directive 98/79/EC on in vitro diagnostic medical devices (IVDD) and, in the absence of an up-to-date replacement, it can still be considered the state of the art.
MEDDEV 2.14/2 states:
“for a product to be categorized as an RUO product it must have no intended medical purpose or objective."
Source: MEDDEV 2.14/2 rev.1
This means that an RUO product must not have even a rudimentary medical purpose.
However, in the case of tests developed in-house by a laboratory (LDTs), this restriction does not apply provided that the products are not sold to other companies. The guidance gives the following specific examples of LDTs that may be designated “research use only” under this requirement:
- PCR enzymes
- Gel component agars
The IVDR also addresses RUO products.
“device for performance study’ means a device intended by the manufacturer to be used in a performance study.
A device intended to be used for research purposes, without any medical objective, shall not be deemed to be a device for performance study; ”
Source: IVDR Art. 2(45)
Thus, the IVDR, like MEDDEV 2.14/1 (IVD Medical Device Borderline and Classification issues), draws a distinction between RUO products and “devices for performance studies.”
Again, the key aspect of the definition is the RUO product’s lack of medical purpose.
To be classed an RUO product, it is vital that the product does not serve a medical purpose. Even a suspected medical purpose is enough for a device to be no longer considered an RUO product.
(See MEDDEV 2.14/1 section 1.1 4.)
Definition in the USA
In 2013, the FDA published a guidance document on RUOs entitled “Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only.”
This guidance defines RUO products as follows:
“ An RUO product is an IVD product that is in the laboratory research phase of development and is being shipped or delivered for an investigation that is not subject to part 812” [NB: Part 812 concerns the provision of devices for performance evaluation purposes as a preliminary step to IVDs]
Source: FDA guidance “Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only”
Some examples of products that the FDA believes fall into this research phase of development are:
- Tests that are in development to identify test kit methodology, necessary components, and analytes to be measured
- Instrumentation, software, or other electrical/mechanical components under development to determine correct settings, subcomponents, subassemblies, basic operational characteristics, and possible use methods
- Reagents under development to determine production methods, purification levels, packaging needs, shelf life, storage conditions, etc.
Therefore, according to the FDA, a clearly visible RUO label must be affixed specifically to products that are in a research phase.
c) What are the consequences of using the “Research Use Only” label?
Normally, IVDs are subject to regulatory requirements (for example, according to the IVDR or FDA) based on their risk class.
However, RUO products do not fall within the definition of “in vitro diagnostic medical devices” given by the IVDR or the relevant FDA regulations . This means that these regulations do not apply to RUO products.
Definition: In vitro diagnostic medical devices (IVDs) in the EU
“‘In vitro diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;”
Source: Article 2 IVDR
Definition: In vitro diagnostic medical devices (IVDs) in the USA
“In vitro diagnostic products are those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.”
Source: 21 CFR 809.3
Therefore, the requirements of the IVDR do not apply to RUO products. In the USA, they are exempt from cGMP and the FDA's quality regulations.
Depending on the product, they may still have to comply with requirements that are not specifically intended for IVDs (such as the REACH regulation for chemicals or the Machinery Directive ).
Since RUO products are thus subject to considerably fewer controls than IVDs, it is necessary to severely restrict their use.
Therefore, in particular they may not be used to:
- Make diagnoses
- Conduct performance studies
2. Use and misuse of “Research Use Only” labels
A) what should ruo products be used for.
As the name “research use only” indicates, products with RUO labeling are intended for research purposes only. RUO products are particularly attractive for the research sector due to the simplified process and lower hurdles for placing them on the market.
MEDDEV. 2.14/2 rev.1 provides a precise list of areas where RUO products may potentially be used:
- Basic research
- Pharmaceutical research
- Better identification and quantification of individual chemical substances or ligands in biological specimens
- In house manufacturing of so called “home brew kits” for research purposes
And of areas where the use of RUOs is expressly not permitted:
- Use of raw materials which are labeled “for “research use only” but which are incorporated into a finished product
- So called “research use” products being tested against a comparator IVD product that bears the CE mark
- Products for market studies/ feasibility studies
b) What RUO products are often used for
However, the low hurdles are also the reason why RUO products are often used for purposes they are not intended for. This poses significant dangers for manufacturers, operators and patients.
Sale of RUO products to medical laboratories
RUO products are sold by manufacturers to medical laboratories. Although doctors sometimes also conduct research, this is not really the main purpose of a medical laboratory. Therefore, when discussing sales with doctors, it should always be assumed that there is a medical reason behind the use of the product.
This means that anyone who knowingly sells RUO products to medical laboratories is potentially under suspicion of using the pretext “for research use only” to ignore an intended medical purpose and thus avoid responsibility for a medical device.
There are certainly laboratory products that clearly have no specific medical purpose, e.g.:
- Nutrient media
- Reaction vessels
- Washing solutions
These products are best labeled as “general laboratory supplies” rather than “RUO”.
Avoid reference to any specific diagnostic procedures in your advertising materials for products that clearly do not have a medical purpose. You should always stay on the technical or purely analytical level.
The issue with analyte specific reagents
Whether an RUO product contains analyte specific reagents, e.g., primary antibodies, FISH probes, PCR primers and probes, and sequencing panels, can be critical. In some cases, a medical purpose can be inferred just from the description of the product's performance.
This would be the case if a manufacturer of a RUO-labeled kit for the detection of viral genes specifies a number of copies per ml of blood that the kit can detect.
ASR in the USA
The FDA abbreviates the term “analyte specific reagents” to “ASR” and defines it as follows:
“Analyte specific reagents (ASR's) are antibodies, both polyclonal and monoclonal, specific receptor proteins, ligands, nucleic acid sequences, and similar reagents which, through specific binding or chemical reaction with substances in a specimen, are intended for use in a diagnostic application for identification and quantification of an individual chemical substance or ligand in biological specimens.”
Source: 21CFR864.4020 a)
In other words, US law says that, by definition, ASRs have a diagnostic purpose.
Exception: The sale of ASRs to IVD manufacturers as components for manufacturing kits or to non-clinical laboratories for research and development without compliance with regulatory requirements is permitted.
ASR in the EU EU law does not contain this exception. Nor does the term “analyte specific reagent” does appear in any of the applicable EU regulations. Therefore, such products may have a general laboratory purpose in the EU, depending on the justification. This means they do not fall under the IVDR if the manufacturer defines the intended purpose accordingly. However, if the manufacturer assigns a medical or diagnostic purpose to these products, the regulatory hurdles will very high once the IVDR comes into full effect (currently scheduled for May 26, 2022).
This means that the crucial factor is whether manufacturers have clearly defined the intended purpose and whether communication with customers (e.g., in advertising materials) is in line with this purpose.
Further information
You can find out more about the intended purpose of medical devices here: Intended purpose and intended use
Use of RUO products in medical laboratories
It is not just manufacturers for whom the sale of RUOs to medical laboratories represents a problem. The laboratories themselves may also not be acting in line with their status as operators and may, as a result, be liable under certain circumstances.
- Medical laboratories are free to develop in-house tests themselves. In such cases, RUO products are often used in diagnostic procedures. Even under the IVDD, MEDDEV 2.14/2 was critical of this. However, with the new In Vitro Diagnostic Medical Device Regulation (IVDR) , the EU is explicitly placing more restrictions on the routine use of such lab developed tests . Read more in our article The EU Is Regulating Medical Laboratories. Are Laboratory Developed Tests Still Allowed? .
- Due to the low regulatory hurdles, purchasing RUO products is very affordable. As a result, medical laboratories prefer them over expensive CE-IVD devices if they can achieve the same level of performance. Nevertheless, the use of RUO products for purposes other than research, even in cases where they provide similar results, is not permitted.

3. Consequences of incorrect classification
Lack of controls can have a negative effect on quality. As a result, the relevant bodies (e.g., authorities during inspections) take a closer look at whether a product is actually intended for “research use only”.
Manufacturers should also be aware that simply sticking an RUO label on a product does not on its own mean that the product no longer has to comply with requirements for IVDs that would otherwise apply.
In its guidance document on RUO , the FDA writes that only the actual intended use qualifies a product as RUO – or doesn’t. The FDA also uses marketing materials or other general factors as evidence of the intended purpose.
"Because these products are exempt from most regulatory controls, it is important that they are not distributed for clinical diagnostic uses. Mere placement of an RUO or IUO label on an IVD product does not render the device exempt from otherwise applicable clearance, approval, or other requirements. FDA may determine that the device is intended for use in clinical diagnosis based on other evidence, including how the device is marketed. ”
Manufacturers and operators who misuse the RUO label could face severe penalties, as such behavior can cause serious harm to patients or even the general public.
a) Consequences for manufacturers and operators
Improperly selling IVDs with an RUO label or using RUO products for purposes other than research is not a trivial offense.
Manufacturers who demonstrably hide or aim to hide a diagnostic purpose behind the RUO label should expect legal consequences in Germany. The same applies for operators who misuse RUO products. There is the possibility of a fine or even prison sentences. In addition, there is potential liability for harm suffered by patients.
b) Consequences in the USA
There are also severe penalties in the USA. If an RUO label is deemed to have been incorrectly used for a product, the product would be considered misbranded under sections 502(a) and 502(o) of 21 US Code, 352(a), 352(o) [A1] and would be considered adulterated under section 501(f) of 21 US Code 351(f).
c) Consequences for patients
However, the consequences can be even worse for patients. After all, the regulatory requirements for IVDs aren’t just plucked out of thin air to annoy manufacturers and operators. The regulations are intended to protect patients against incorrect results and subsequent wrong decisions. False-negative results can lull patients into a false sense of security and an existing disease may worsen undetected. One example would be the metastasis of an undetected cancer due to a test not performing as intended.
Some incorrect diagnoses could even be so severe that they can cause the death of a lot of people: an undetected viral infection can cost many lives in the early stages of an epidemic or pandemic, as the coronavirus pandemic sadly demonstrated.
4. Alternatives to “research use only” products
To avoid legal problems and risks for third parties, manufacturers and users should use alternatives to RUO products in borderline cases.
These alternatives don’t always have to be CE-IVDs. Depending on the specific situation, the following alternatives can be considered based on the intended purpose:
a) Products for general laboratory use
According to the MEDDEV 2.14/1 (IVD Medical Device Borderline and Classification Issues) guidance, it is a product's characteristics that determine whether it can be classified as a product for general laboratory use or not.
- If, based on its characteristics, a product is not specifically intended to be used for in vitro diagnostic examinations, it is not an IVD.
- Manufacturers cannot label products for general laboratory use as IVDs.
RUO products used for a better identification and quantification of individual chemical substances or ligands in biological specimens
Source: MEDDEV 2.14/2
Such products must have a general use. However, use as an IVD does not have to be ruled out, provided the product is not made specifically for a particular test. According to MEDDEV 2.14/2, even the aforementioned analyte specific reagents (ASRs) without a medical purpose fall into this category.
There are several advantages to using products for general laboratory use instead of RUO products:
- The product does not fall under the IVD Directive or the IVDR, which saves you a lot of time and money.
- Laboratories that use these products for in-house procedures are not in danger of being accused of using RUO products in routine diagnostic procedures.
However, the disadvantage is that the medical laboratory is responsible for ensuring that the examination conforms with the IVDR. This can make the product less interesting because the regulatory requirements entail a lot of work.
b) Lab developed tests with class A CE-IVDs Manufacturers may sell general laboratory reagents, which can be authorized as IVDs under the IVDR, to medical laboratories.
In combination with the ASRs developed in-house, laboratories can validate and use these products as lab developed tests (LDTs).
Read our article on lab developed tests to find out what laboratories should be aware of.
c) “For performance evaluation only” as a preliminary stage for certified IVDs
The IVDR defines " device for performance studies ” as follows:
“‘Device for performance study’ means a device intended by the manufacturer to be used in a performance study.”
Source: IVDR 2017/746/EU
These devices must already be safe, as far as possible, and meet the relevant general safety and performance requirements.
5. Ways to protect yourself
Manufacturers, operators and patients can take the following steps to avoid legal and other negative consequences when using RUO products:
a) Manufacturers
In the case of manufacturers, it is particularly important that they narrowly define the intended purpose of their product.
Analyte specific reagents should only be labeled as RUO products for specific non-medical purposes.
Example: SARS-CoV-2 and its mutations: a test kit that uses specific primers and probes to distinguish the variants B.1.1.7 (alpha variant) and B.1.351 (beta variant) from the initial variant following a positive result may be an RUO product if it is only intended to be used to determine the prevalence of the variant in the population. A specific intended purpose in this case would be: “Intended solely for epidemiological research for the purpose of surveying the prevalence of SARS-CoV-2 variants in the general population.” If a medical laboratory subsequently, based on new findings, used this test to provide the best possible treatment for infection by a specific variant, this would be an off-label use. The laboratory would then be responsible for the test's conformity.
Provided the manufacturer did not advertise the product with this clinical benefit, it would be adequately protected.
b) Operators
Operators should record exactly what they use IVDs and RUO products for.
Medical laboratories are operators of medical devices and IVDs and, therefore, are responsible for only using medical devices according to their intended purpose and in accordance with the generally accepted rules of the technology. This is stipulated in Section 4 of the German Medizinprodukte-Betreiberverordnung (MPBetreibV (German)). To be on the safe side, laboratories should keep a record of which medical devices and IVDs are in operation and routine use. This record should include a reference to the applicable test procedure and the intended purpose of the IVD.
This record can also be used to identify investigational procedures for which there are no adequate CE-IVDs available on the market. The lack of alternatives would justify the use of RUOs (as lab developed tests) in validated processes it has developed in-house, provided that the laboratory checks and can demonstrate that the general safety and performance requirements and the additional requirements of Article 5(5) of the IVDR are met.
Read more about the requirements for LDTs in our article on the topic .
c) Patients
Patients lack the knowledge to recognize what is and isn’t an RUO on their own. They are often given little to no information about the test they are undergoing. So, patients should follow this basic rule: ask your doctor or pharmacist!
- Patients can ask for the complete test report from the laboratory so that they can get a second opinion in case of doubt. The report should also indicate which specific test was performed.
- Patients should inform themselves about how “well” or “poorly” a test works, as well as the benefit-risk ratio.
- In the future, patients and doctors will also be able to get information about medical devices from EUDAMED and use this information to decide whether or not the test was performed with certified and thus legally compliant IVDs.
6. Conclusion
In the opinion of the EU Commission and the FDA, products “for research use only" have no place in diagnostics. To be used for diagnostic purposes, products have to go through the necessary controls. But these controls do not apply to RUO products.
Anyone who ignores this prohibition and uses or sells RUO products for purposes other than pure research is playing with fire. Manufacturers and operators run the risk of legal trouble and could even endanger patients’ health. Therefore, RUO products should only be used for research purposes. For other uses, manufacturers and operators should use the alternatives mentioned.
Our tip is: if you, as a manufacturer or medical laboratory, find that an RUO product is particularly well-suited for in vitro diagnostics, consider whether further development and conformity assessment to make it an IVD is worthwhile. We will be happy to help you work out which of the three alternatives to RUOs mentioned above is the best alternative to your product as part of our IVD authorization strategy consultation. If necessary, we can also help you ensure your product development conforms with the regulations.

Dr. Diana Gabriel

A quick overview: Our
Starter-Kit
Always up to date: Our
Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.
Individual Cookie Settings
Only accept required cookies.
Privacy Notes Imprint
Here is an overview of all cookies use
Required Cookies
These cookies are needed to let the basic page functionallity work correctly.
Show Cookie Informationen
Hide Cookie Information
Provide load balancing functionality.
Provides functions across pages.
Hubspot Forms
Used for the google recaptcha verification for online forms.
Cookies for Statistics
Statistic cookies anonymize your data and use it. These information will help us to learn, how the users are using our website.
Google Analytics
Tracking and analys of traffic on our websites.
Cookies for Marketing
Marketing cookies from thrid parties will be used to show personal advertisment. They use them to track users outside of their own web page.
Keeping track of a visitor's identity. It is passed to HubSpot on form submission and used when deduplicating contacts. It contains an opaque GUID to represent the current visitor. It also introduces cookies from linked in for marketing reasons.
LinkedIn conversion tracking.
Cookies for external Content
Content for Videoplatforms und Social Media Platforms will be disabled automaticly. To see content from external sources, you need to enable it in the cookie settings.
Google Maps
Used to display google maps on our Websites. Google uses cookies to identify and track users.
An Introduction to Research Use Only (RUO)

In this blog, we recap our eBook, “An Introduction to Research Use Only (RUO)” – Click HERE to download the entire publication.
Learn how it differs from adjacent labels, the FDA and EU guidance, its appropriate use, and the consequences of mislabeling products RUO.
Introduction
In the complex world of medical device development, regulation, and distribution, finding the appropriate label to put on a device may not be simple. When is one label appropriate over another? Does a device need to go through additional testing, verification, or validation? And what are the consequences of using the wrong label? In this eBook, we’ll cover the differences between Research Use Only (RUO) and a medical device – although, it’s generally a very clear distinction.
Using the right language and label is critical to complying with best practices. This is why Regulatory Affairs works with the regulatory bodies to ensure that the limitations of the product are properly documented. In a rush to get products to market, it may be tempting to use a Research Use Only (RUO) label to avoid additional regulatory processes while still empowering other researchers and developers. However, there are risks to using the RUO label inappropriately that can have serious consequences for developers, users, and patients. In fact, mislabeling a product is illegal, and punishable. You can see an example warning letter the FDA sent to Carolina Liquid Chemistries Corp after finding intentional mislabeling in 2019 here.
This introduction will provide an overview of the Research Use Only label, how it differs from similar, adjacent labels, its appropriate use, and the consequences of mislabeling products RUO.
What is Research Use Only (RUO)?
The label Research Use Only (RUO) is generally used to indicate products that are intended for scientific research only. They cannot be used for diagnostic or medical purposes. However, there is no standard definition of “research use only,” and the label has slightly different meanings in the European Union and the United States. With the IVDR regulations, RUO products that are being used in the LDT space are going to be revisited and potentially reclassified as a medical device. With this new classification, teams will likely need to follow design controls, best practices, and industry standards.
What is the FDA guidance on Research Use Only products?
Under the FDA’s guidance issued in 2013 , a product labeled Research Use Only is an In Vitro Diagnostic (IVD) product “that is in the laboratory research phase of development and is being shipped or delivered for an investigation that is not subject to part 812.” The agency includes in this category:
- “Tests that are in development to identify test kit methodology, necessary components, and analytes to be measured.
- “Instrumentation, software, or other electrical/mechanical components under development to determine correct settings, subcomponents, subassemblies, basic operational characteristics, and possible use methods.
- “Reagents under development to determine production methods, purification levels, packaging needs, shelf life, storage conditions, etc.”
The European guidance document MEDDEV 2.14/2 states that a product categorized as an RUO product “must have no intended medical purpose or objective.” The guidance does exempt some tests developed for in-house use as long as the products are not sold to other companies. Some examples of items that can be classified as “research use only” under this exemption include PCR enzymes, gel component agars, and primers.
RELATED: FDA released new draft guidance of premarket submissions for medical devices – are you ready?
What is the difference between ruo and ivd.
An IVD is an “In Vitro Diagnostic Medical Device,” and the general term applies to any device or product that either alone or with other products is intended to be used for diagnostic, monitoring, or compatibility purposes. There are four different regulatory levels for IVDs:
- Research Use Only (RUO)
- General Laboratory Use (GLU)
- For Performance Studies Only (PSO)
- In Vitro Diagnostic Medical Device (IVD)

The simplest explanation for these different levels is that each increasing level requires more testing and oversight. Research Use Only products are at the lowest level of regulation, and In Vitro Diagnostic Medical Devices are at the highest level. Occasionally in the US, products will be labeled as “RUO IVD,” which means an in vitro device that is intended for research use only.
Products labeled with the “CE-IVD” label indicate that they have progressed through the applicable regulatory process and standards (such as IVDD or IVDR). These products are approved for diagnostic use and must include the IVD symbol to be used for medical purposes.
In the EU, as of May 2022, IVDs must comply with Regulation (EU) 2017/746 (IVDR) . The IVDR defines IVDs as follows:
“‘in vitro diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state; (b) concerning congenital physical or mental impairments; (c) concerning the predisposition to a medical condition or a disease; (d) to determine the safety and compatibility with potential recipients; (e) to predict treatment response or reactions; (f) to define or monitoring therapeutic measures.”
All IVDs that comply with the IVDR must carry the CE Mark if marketed in the EU.
Research Use Only products are not subject to regulatory requirements in either the US or the EU, but because they don’t meet the same compliance standards as IVDs, they must be clearly labeled as RUO products and cannot be used for medical purposes.
A known exception is the lab developed test (LDT) pathway for clinical purposes.
What are the requirements for an RUO product?
In the US, RUO products are basically unregulated and do not need to meet any specific requirements to carry the RUO label. The FDA does not specify any restrictions or limitations on RUO products, provided they are clearly labeled “For Research Use Only. Not for use in diagnostic procedures.” For this reason, RUO products can be an excellent solution for laboratories that need research materials for testing and research purposes. Because products with the RUO label do not require extensive testing, verification, and validation, they tend to be more cost-effective for research purposes.
The EU rules are similar. Because RUO products do not have clinical applications, they are not considered medical devices, and there are no requirements for RUO products defined by either the IVDD or the IVDR. These products should not be marked with the IVD mark, and they should be clearly labeled as “Research Use Only.”
RELATED: See how Jama Software ® helped Össur improve the mobility of millions by replacing process rigidity with speed and agility.
Are there alternatives to ruo labels.
Given the significant differences between labeling a product as RUO and labeling a product as IVD, manufacturers and users can’t be too careful when it comes to assigning labels or using products for specific purposes. If there is a risk to using products labeled as RUO, manufacturers and users should opt for products that have attained a higher compliance level. For example, for a doctor’s office or home use, IVD is the right path. For clinical purposes or hospital labs, RUO could be used as LDT as long as they are CAP/CLIA certified, such was the case with COVID-19 testing kits when the pandemic first hit.
For products that meet a higher degree of compliance, it is possible to assign General Laboratory Use (GLU), Performance Studies Only (PSO), or even In Vitro Diagnostic Medical Device (IVD) labels. However, depending on the intended use for the Research Use Only products, pursuing these additional levels of compliance may or may not make sense.
What is CLIA certification?
CLIA stands for Clinical Laboratory Improvement Amendments. The Centers for Medicare & Medicaid Services (CMS) regulates all clinical laboratory testing performed on humans in the United States through CLIA.
What is a CAP accreditation?
CAP stands for The College of American Pathologists (CAP) . The purpose of CAP laboratory accreditation is to ensure laboratories provide precise test results for accurate patient diagnoses, meet CLIA and CAP requirements, and demonstrate compliance with professionally and scientifically sound and approved laboratory operating standards.
What are RUO products used for?
As the name implies, RUO projects should be used for research purposes only. They may be used for basic research, pharmaceutical research, or in-house manufacturing of “home brew kits” for research purposes and potentially for clinical applications via the LDT pathway. RUO products are specifically not to be used to make diagnoses, conduct performance studies, or as a substitute or comparator for a CE-IVD device. They may also not be used for market or feasibility studies. Raw ingredients labeled as RUO products may not be incorporated into a finished IVD product.
Learn more about the advantages and disadvantages of the RUO label (and more) by downloading the entire eBook HERE .
- Recent Posts
- [Webinar Recap] Key Systems Engineering Skills: Critical Thinking and Problem Framing - March 5, 2024
- Jama Connect® Features in Five: Medical Device & Life Sciences Solution 2.0 – Part 2 - July 28, 2023
- Jama Connect® Features in Five: Medical Device & Life Sciences Solution 2.0 – Part 1 - July 21, 2023
USA 135 SW Taylor Suite 200 Portland, Oregon, 97204
EUROPE Amsterdam Queens Tower Delflandlaan 1, 1062EA Amsterdam The Netherlands
© 2024 Jama Software
- JAMA CONNECT
- Product Overview
- Pricing and Licensing
- Success Programs
- Resource Library
- User Community
- Privacy Policy
- Privacy and Security
- Preferences
We use cookies to offer you a better browsing experience and analyse our website performance. We also use third-party cookies to further customise your experience showing relevant content while you are navigating on third-party platforms.
Our Priorities MedTech Europe strives to support our dynamic sector in meeting the needs of patients and health systems. To achieve this, we focus on engaging with healthcare stakeholders on key issues from regulations and market access to digital health and Brexit, among others.
- COVID-19 Information Hub
- Interactions with the Medical Community
- Access to Medical Technology
- Medical Technology Regulations
- Digital Health
- International
- Environmental and Social Sustainability
- Market Data
- Research and Innovation
- Innovative Health Initiative (IHI)
Sector Groups MedTech Europe sector groups bring together company experts to drive forward key healthcare domains, helping to address issues facing these sectors and shaping their future. We have dedicated groups focused on cardiovascular health, ophthalmology, diabetes, orthopaedics, and AMR/HAI.
- Antimicrobial Resistance (AMR) and Healthcare Associated Infections (HAIS)
- Cardiovascular
- Homecare & Community Care
- Orthopaedic
Real stories of people’s lives transformed by medtech.
Your platform for dialogue about medical technologies.
Search on this website
Research Use Only Products

What are Research Use Only (RUO) products? Research Use Only (RUO) products are a distinct category of in vitro diagnostics (IVDs) exclusively tailored for laboratory research. RUOs encompass specialised reagents, equipment, and materials crucial for scientific investigations, contributing significantly to the development of cutting-edge tools and solutions for research applications.
Research Use Only (RUO) products play a crucial role in medical research and innovative management of many patients. These specialised products, which include laboratory reagents and equipment, are exclusively designed for research in controlled laboratory environments. As essential tools for medical and scientific investigations, experimentation, and analysis, RUOs contribute to developing innovative solutions and advancements in medical research.
For example: RUO products can be used for Fundamental Research, in Pharmaceutical Research to find new drug compounds, and for a better identification and quantification of individual chemical substances. In diagnostics research, RUO products are essential to the development of new diagnostic assays and tools.
Unlike in vitro diagnostic medical devices (IVDs), RUOs are dedicated to facilitating research initiatives and are not intended for direct medical procedures with human patients. RUOs are not defined in the EU’s In Vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR); they are regulated by the EU General Product Safety Regulation and other applicable EU legislations. Manufacturers of RUO products clearly label them as “Research Use Only” and use the RUO label.
From a production and specifications general perspective, the knowledge and processes needed to manufacture RUOs are very similar to those needed to manufacture CE marked IVDs. Many companies which operate in the IVD space will have RUO products in their portfolio. RUOs will generally have a similar chemical and physical composition compared to IVDs, but their intended purpose will be different. While RUO or IVDs might seem similar in their appearance and specifications, unambiguous and documented evidence associating the use of devices with in vitro diagnostic examination procedures is required to qualify a device as an IVD.
RUOs provide researchers and scientists – including those operating in medical laboratories – with valuable resources to advance in the understanding of disease, in drug discovery, in the development of new therapies and diagnostic tools. Laboratories or research consortia often collaborate with RUO manufacturers to tailor products to meet specific research needs and requirements, fostering a collaborative environment and contributing to the continuous evolution of research tools and solutions.
One critical application of RUO is to enable medical laboratories to develop in-house assays to e.g. diagnose rare and emerging conditions or to improve the current knowledge and management of specific diseases for which no adequate CE marked IVDs exist. This not only fulfils a critical and imminent healthcare need but is also a key stepping stone in the eventual development of IVDs. A poignant example of this was the development of COVID-19 assays during the early phase of the pandemic – initially, reference laboratories developed in house assays test for the SARS-CoV-2 virus, and shortly afterwards, commercial IVDs began to reach the market in order to fulfil a critical need during the global health crisis. However, it is worth noting that the use of in-house assays is regulated in IVDR and is subject to certain conditions.
In essence, RUO products provide researchers and physicians with the necessary tools to conduct experiments and studies, contributing to the overall progress in medical research. Their intended use in laboratory settings supports the development of new technologies and innovative solutions for various research applications.
Share this page
MedTech Europe Manifesto for 2024 – 2029
Empowering Patients, Inspiring Innovation
Sign up for your monthly newsletter
By clicking the Subscribe button, you give consent to MedTech Europe AISBL to use of the information you provided and send you content on the services you selected. We will ensure that the information is processed confidentially, and will only share it with third party providers that assist in providing these services. These providers may be located outside the EU; in this case, we will ensure that they are subject to a legal framework adequate in safeguarding your data, in compliance with European data protection law. You can unsubscribe, change your preferences or update your information at any time by clicking on the unsubscribe button available on all messages. For more information on how MedTech Europe will handle your personal data, please refer to our Privacy Policy . You can contact us at [email protected] for further questions related to your privacy and your rights.
- Featured News
- Artificial Intelligence
- Bioprocessing
- Drug Discovery
- Genome Editing
- Infectious Diseases
- Translational Medicine
- Browse Issues
- Learning Labs
- eBooks/Perspectives
- GEN Biotechnology
- Re:Gen Open
- New Products
- Conference Calendar
- Get GEN Magazine
- Get GEN eNewsletters
Oversight of Research Use Only Products
By Jeffrey N. Gibbs
March 1, 2010 (Vol. 30, No. 5)
RUO Assays and Instruments Face Greater Scrutiny
The FDA actively regulates medical devices intended for diagnostic use. Diagnostic kits intended for diagnostic use face the full panoply of FDA regulation. In sharp contrast, research use only (RUO) products are essentially unregulated. In fact, although RUO products are often discussed as though they are a kind of medical device, RUOs are not devices at all.
A commercially important class of products, RUOs are defined very briefly by FDA regulations. RUO products are described as products “in the laboratory research phase of development and not represented as an effective in vitro diagnostic product.” This definition has created some uncertainty as to what products fall into the RUO category.
The same regulation establishing the RUO category requires that RUO products bear the following labeling statement: “For Research Use Only—Not for use in diagnostic procedures.” Although not authorized by the regulation, many companies have shortened the statement to just the first clause. FDA regulations do not prescribe any other restrictions or limitations on RUO products beyond this labeling statement. Thus, FDA regulations define the category and prescribe labeling, and nothing more.
Given that RUO products are not intended to diagnose “a disease or other condition,” it is not clear that they are even subject to FDA’s jurisdiction. The intended use of an RUO product—research, not diagnosis—presumptively removes it from the definition of a device and FDA’s authority.
In any event, aside from bearing the mandated statement, RUO products are not regulated by the agency. For example, they do not need to be listed with FDA or comply with the Quality System Regulation (QSR). They can be sold without any FDA clearance or approval. As a practical matter an RUO is essentially unregulated by FDA.
Over the years, the paramount regulatory issue for products bearing the RUO label has been whether or not they actually do belong within the RUO category. There have been multiple instances in which RUO products have become widely used by laboratories for clinical applications. There have also been a number of occasions where companies have labeled products as RUO but then promoted them for diagnostic use. In some instances, companies have made specific diagnostic claims for their assay or instrument but still labeled the product as RUO.
Biomarker kits are often labeled as RUO because it is not known whether the product has any clinical use or, if so, what that use might be. The assay’s developer may expect that a particular biological substance will be of some clinical value, but not be sure what that value is. Labeling a product RUO, allows it to get into the hands of researchers who can then evaluate whether the product may be potentially valuable for some specific diagnostic purpose.
Often, no clinical use is ever identified. Some assays maintain their true RUO status indefinitely. While the product may be helpful to researchers in understanding basic biological mechanisms, a diagnostic use may never be discovered.

Guidance Documents
FDA has initiated several attempts to try to regulate RUO products more tightly. In the early 1990s, FDA issued a draft Compliance Policy Guide (CPG) document that sought to significantly restrict the availability of RUO products. This guidance document went through several iterations but was never finalized. There is still no guidance document setting out FDA’s policy regarding RUO products, however, reports have recently surfaced that a new RUO policy may finally be released.
One of the elements set forth in the draft CPG was that the distributor of the RUO product should receive a certification from the laboratory customer that the product will be used for research purposes only. Although the CPG was not adopted, some vendors have asked laboratories to sign some type of acknowledgement form. While this will help support a vendor’s position that its product is intended only for research use, it is not currently required. FDA has, however, “encouraged” some instrument suppliers to adopt certification programs.
Concerned by the proliferation of RUO products, in 1997 FDA tried a different tack. That year, FDA promulgated the Analyte Specific Reagent (ASR) regulation. ASRs were broadly defined as the building blocks of diagnostic assays. Unlike RUOs, ASRs were subject to FDA requirements, including QSRs and Medical Device Reporting. This regulation was prompted, in part, by the belief that it would result in the availability of higher quality materials for laboratory tests and displace some of the lower quality RUOs.
To some degree, that plan succeeded. Many different products were offered to laboratories as ASRs. However, while many of these were basic chemical components, more complex products were also sold as ASRs. Ultimately, FDA concluded that the ASR regulation was being used as a vehicle for products that didn’t fit the intent of the regulation.
FDA therefore released a guidance document in 2007 that substantially curbed the availability of ASRs by prohibiting companies from combining more than one active component. With the advent of molecular diagnostics, selling a single component was often impracticable, e.g., a primer and probe pair need to be offered together. This narrow interpretation of ASRs has essentially precluded the sale of ASRs for use in molecular diagnostics. Somewhat predictability, a number of companies responded by relabeling their ASRs as RUOs. This has helped lead to a renewed focus on RUOs by FDA.
For years, the principal regulatory question for products labeled as RUOs has been whether they qualify for this classification and hence are not subject to regulation as devices. While FDA has not issued either a regulation or guidance delineating how companies can promote RUOs, the agency has taken enforcement action against a number of RUO companies.
Even absent regulations or guidance, it is apparent that in FDA’s view a product forfeits its RUO status if certain types of claims are made—claims that the product can diagnose a disease or condition, provide clinical sensitivity or specificity data, or offers a clinical benefit. Correspondingly, the instructions for use (IFU) accompanying the product need to be brief.
While the bulk of RUO products have been assays, the RUO category also encompasses instruments and equipment. This can present its own set of regulatory challenges, particularly when an IVD applicant has used an RUO instrument in conjunction with developing its assay, a situation that is now occurring with greater frequency.
The utilization of RUO instruments in assay development has led to the submission of applications that reference RUO instruments. This may result in naming the RUO instrument in the draft IFU, i.e., the applicant states that the assay is to be performed on an RUO instrument, or the data for the IVD were generated on an RUO instrument.
While FDA had accepted these practices, that has seemingly changed. Therefore, an IVD company that has tested and validated its assay on an RUO instrument or is using RUO assays in its test system should discuss with FDA at an early stage how to address the regulatory implications that may arise from this situation. Simultaneously, companies that are selling RUO-labeled instruments that are being widely used in diagnostics may find that they will be receiving more regulatory scrutiny from FDA.
Over the past few years, RUO products have received relatively little attention from FDA. That regulatory lull seems to be ending.
Jeffrey N. Gibbs ( [email protected] ) is a director at Hyman, Phelps & McNamara. Web: www.hpm.com.
Single-Cell Cloning Remains a Challenge
New approach to nanopore sequencing that is sure to catch your....
- Search Menu
- Sign in through your institution
- Advance Articles
- Clinical Case Studies
- Journal Club
- Clinical Chemistry Podcasts
- Clinical Trainee Council
- Special Issues
- Clinical Chemistry Guide to Scientific Writing
- Clinical Chemistry Guide to Manuscript Review
- Author Guidelines
- Submission Site
- Self-Archiving Policy
- Call for Papers
- Why Publish?
- About Clinical Chemistry
- Editorial Board
- Advertising & Corporate Services
- Journals on Oxford Academic
- Books on Oxford Academic
Article Contents
2 nonstandard abbreviations:.
- < Previous
“Research Use Only” Reagents: Is There an Imperative for Increased FDA Oversight?
- Article contents
- Figures & tables
- Supplementary Data
Timothy J O'Leary, “Research Use Only” Reagents: Is There an Imperative for Increased FDA Oversight?, Clinical Chemistry , Volume 57, Issue 12, 1 December 2011, Pages 1681–1683, https://doi.org/10.1373/clinchem.2011.174268
- Permissions Icon Permissions
On June 1, 2011, the US Food and Drug Administration (FDA) 2 Office of In Vitro Diagnostic Device Evaluation and Safety issued draft guidance for industry and FDA staff intended to provide guidance regarding the FDA's thinking about in vitro diagnostic device (IVD) products labeled for “Research Use Only” (RUO) or “Investigational Use Only” (IUO) ( 1 ). Reagents may be marketed under either of these labels without FDA premarket review and are partially or totally exempt from compliance with the Quality Systems Regulation (21 CFR 820). Therefore, an IVD manufacturer might it very tempting to avoid the trouble and expense of a 510(k) or premarket-approval submission by labeling a product as RUO or IUO, despite knowing full well that the product is likely being used as an IVD under conditions in which there is no research protocol and no oversight by an institutional review board.
There is no doubt that the FDA's intent is to improve patient safety. Compliance with the Quality Systems Regulation provides assurance that an IVD meets manufacturing specifications and that lot-to-lot variations are minimized and well understood. FDA clearance provides further assurance that the performance characteristics of laboratory tests are sufficiently well understood as to enable their intelligent use. Furthermore, the use by clinical laboratories of FDA-cleared IVD products likely reduces interlaboratory variation in testing and increases the ease with which results can be “ported” from one healthcare facility to another, potentially reducing healthcare costs that arise from duplicate testing. Nevertheless, the laboratory community is right to be concerned about the guidance document statement that manufacturers should not sell RUO or IUO reagents “to laboratories that they know use the product for clinical diagnostics use.” This statement could have unintended consequences that adversely affect patient care. In particular, the molecular diagnostics community has expressed concern that such reagents as PCR primers and sequencing reagents and equipment could become unavailable and that this outcome could affect many aspects of medical care, including newborn screening, HLA testing, and human papilloma virus genotyping, among others.
When considering approaches by which the FDA and the laboratory community can improve patient care and safety, it is important to consider the overall medical and regulatory environment in which laboratory tests that currently use RUO and IOU IVDs are conducted. In the remainder of this Opinion, I consider potential deficits in laboratory testing that currently do not rely on FDA-cleared IVDs and discuss principles that the FDA and other agencies may wish to consider when determining whether a regulatory solution is the most appropriate way to address the perceived deficits in laboratory testing.
The design and implementation of regulations entail substantial costs on the part of both regulator and regulated, both of which must ultimately be borne by the public. Therefore, regulators should take a measured approach when deciding to implement a regulatory solution to a perceived problem. In my opinion, such decisions should be based on a careful and narrow definition of the public health problem to be addressed, a scientifically valid and quantitative assessment of the magnitude of this problem, and strong evidence that the overall cost of implementing the regulatory approach (including both the cost to the regulator and the cost to the regulated entities) is cost-effective. The regulatory reasoning and the cost–benefit analysis should be published together to facilitate public scrutiny and comment. Implementation of this approach would unquestionably be associated, at least initially, with an adverse financial impact on regulatory agencies, because the assessment of regulatory impact would undoubtedly be more costly than is currently the case. The overall cost to the public seems likely to be offset, at least in part, by avoidance and/or rescission of ineffective and costly regulatory interventions. The recent FDA guidance document does not implement such an approach, so I consider some of the issues that the agency may have attempted to address.
Several potential problems are associated with the use of RUO and IUO reagents in clinical laboratory testing: ( a ) the creation of an uneven playing field for manufacturers; ( b ) the perception that manufacturers or laboratories are defying FDA regulatory authority; and ( c ) the reporting of inaccurate, misleading, or inconsistent results, either within a laboratory or among several laboratories. There is no question that the cost avoidance produced by ignoring regulatory requirements creates an economic environment that gives unfair advantage to commercial manufacturers, compared with institutions that play by the FDA interpretation of the law. Both the clinical investigations and the paperwork requirements associated with FDA submissions are costly. These costs may come at the expense of profit or at the expense of healthcare organizations and insurers (including the federal government).
Inaccurate or misleading results can occur when a clinical laboratory result does not mean what a clinician believes that it means. That situation could arise, for example, if an RUO or IUO reagent is not what the vendor says it is or performs in a manner that both is inconsistent with what the vendor states and is unexpected by the clinical laboratory. It is thus incumbent on laboratories to conduct their quality-assurance activities in a manner that ensures that reported results mean exactly what they purport to mean, whether or not a laboratory test has been cleared by the FDA. Under the framework proposed above, increased regulatory effort by the FDA might be appropriate if evidence of harm exists, although CLIA also provides a framework for achieving this objective.
Although the FDA has not officially elucidated the reason for issuing the guidance document, there have been a number of reports of inaccurate testing with laboratory-developed tests. Perhaps the most prominent is prescribing Herceptin® based on the results of immunohistochemical tests for HER2 (human epidermal growth factor receptor 2) overexpression. 3 Some of these results have depended on the use of uncalibrated laboratory-developed tests; indeed, some authors have postulated that as much as 20% of immunohistochemical HER2 testing used for selecting patients for Herceptin therapy has not been correlated, either directly or indirectly, with response to the drug ( 2 ). If true, that finding demonstrates a clear failure of the CLIA framework alone to adequately protect public health, and it seems likely that the burden of regulatory compliance, if narrowly directed to this issue, is proportionate to the problem. Inaccurate or misleading results have also been associated with direct-to-consumer genetic testing ( 3 ), which is widespread. The magnitude of the harm associated with such testing is uncertain but may be considerable, and oversight under the CLIA mechanism has failed to address the issue.
The potential reach of the FDA guidance document is broader than necessary to deal with HER2 assessment and direct-to-consumer testing. There are substantial numbers of important laboratory-developed tests for which only RUO and IUO reagents are currently available. The FDA guidance document (which, I should note, is nonbinding) could lead to withdrawal of these tests from clinical practice. In my opinion, FDA officials should work to minimize the likelihood of such an occurrence. In addition, there is a risk that innovative new tests for which only RUO and IUO reagents are available will not be deployed in a timely way, further compromising patient well-being. There is clearly a role in medical practice for laboratory-developed and -validated tests that will, of necessity, use reagents that have not passed FDA muster. The failure of CLIA oversight mechanisms is not a reason for the FDA to act unilaterally. Enforcement of both the Food Drug and Cosmetics Act and the Public Health Service Act [which created the CLIA 88 (CLIA amendments of 1988) framework] falls to the Department of Health and Human Services. It is thus appropriate for the FDA and the Centers for Medicare and Medicaid Services to work cooperatively, not only with each other but also with industry and laboratory communities, to develop a robust framework for reducing inaccurate and unreliable laboratory testing while maintaining access to high-quality laboratory testing and minimizing its economic burden.
US Food and Drug Administration
in vitro diagnostic device
Research Use Only
Investigational Use Only
human epidermal growth factor receptor 2
CLIA amendments of 1988.
HER2 is used in this Opinion to refer to the protein encoded by the gene with HUGO-approved gene symbol ERBB2 [v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian)], as HER2 is the name commonly used in practice.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article .
Authors' Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: T.J. O'Leary, Association for Molecular Pathology and Journal of Molecular Diagnostics .
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Research Funding: None declared.
Expert Testimony: None declared.
Disclaimer: The views or opinions expressed in this paper are those of the author and are not to be construed as official or as representing the views of the Department of Veterans Affairs or any other entity of the United States government.
U.S. Food and Drug Administration . Draft guidance for industry and FDA staff-commercially distributed in vitro diagnostic products labeled for research use only or investigational use only: frequently asked questions . http://www.fda.gov/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm253307.htm (Accessed October 2011) .
Carlson B . HER2 tests: How do we choose? Biotechnol Healthc 2008 ; 5 : 23 – 7 .
Google Scholar
U.S. Government Accountability Office . Direct-to-consumer genetic tests: misleading test results are further complicated by deceptive marketing and other questionable practices . http://www.gao.gov/new.items/d10847t.pdf (Accessed October 2011) .
Email alerts
Citing articles via.
- Recommend to Your Librarian
- Advertising and Corporate Services
- Journals Career Network
Affiliations
- Online ISSN 1530-8561
- Print ISSN 0009-9147
- Copyright © 2024 Association for Diagnostics & Laboratory Medicine
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
- Skip to main content
- Skip to FDA Search
- Skip to in this section menu
- Skip to footer links

The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
U.S. Food and Drug Administration
- Search
- Menu
- Medical Devices
- Device Advice: Comprehensive Regulatory Assistance
- Overview of Device Regulation
- Device Labeling
In Vitro Diagnostic Device Labeling Requirements
On this page: , introduction.
- Label Requirements for the Immediate Container
- Labeling Requirements for Inserts and Outer packaging
Exemptions from Labeling Requirements
- Specific labeling requirements for analyte specific reagents
- Restrictions on sale distribution and use of analyte specific reagents
- Specific labeling requirements for OTC test sample collection systems
- Restrictions on the sale, distribution and use of OTC test sample collection systems
In vitro diagnostic products (IVD's) are those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body. In vitro diagnostic (IVD) labeling requirements are located in 21 CFR Part 809. Numbers appearing in parentheses next to subject headings are the corresponding sections of 21 CFR. This section contains the basis requirements for label and labeling (package insert) as specified in the labeling regulations for in vitro diagnostic products.
Label Requirements for the Immediate Container [ 21 CFR 809.10 (a)]
The label for IVD's must state the following information, except in cases where it is not applicable. In addition, all information must appear on the outside container or wrapper, or be easily legible through the outside container or wrapper.
If the presence of any label information will interfere with the test, the information may appear on the outside wrapper or container instead of the label.
If the immediate containers are too small, or otherwise unable to bear labels with sufficient space, then the required labeling as listed below annotated with an asterisk (*) may appear on the outer container labeling only.
Label requirements are as follows:
- The established and proprietary names of the product, e.g., cholesterol meters;
- * The intended use or uses, e.g., pregnancy detection, diabetes screening, etc.;
- A statement of warnings or precautions for users listed in 16 CFR part 1500 (hazardous substances) and any other warnings appropriate to user hazards, and a statement "For In Vitro Diagnostic Use;"
- Name and place of business of the manufacturer, packer, or distributor;
- - Multiple unit products must have traceability of the individual units;
- - Instrument lot numbers must allow for traceability of subassemblies; and
- - A multiple unit product that requires use of its components as a system should have the same lot number, or other suitable uniform identification, on all units.
- - Established (common or usual) name;
- - Quantity, proportion, or concentration of all active ingredients; e.g., mg., weight per unit volume, mg./dl etc., and for reagents derived from biological materials the source and measure of its activity, e.g., bovine, I.U., etc.;
- - Storage instructions adequate to protect the stability of the product, i.e., temperature, humidity, etc.;
- - Instructions for manipulation of products requiring mixing or reconstitution, along with instructions for storage of products that have been reconstituted or mixed;
- i. expiration date (date beyond which the product is not to be used);
- * ii. statement of any visual indication of alteration;
- * iii. Instructions for a simple check to assure product usefulness;
- * - The net quantity of contents.
Labeling Requirements for Inserts and Outer Packaging 21 CFR 809.10 (b)
Labeling must contain in one place the following information in the FORMAT and ORDER listed below, except where information is not applicable, or as specified in a standard for a particular product class.
If the device is a reagent intended as a replacement in a diagnostic system, labeling may be limited to that information necessary to adequately identify the reagent and to describe its use in the system.
If the device is a multiple purpose instrument used for diagnostic purposes, and not committed to specific diagnostic procedures or systems, labeling can be restricted to those points annotated by an asterisk (*).
- * The proprietary and established product name;
- * The intended use of the product and whether it is a qualitative or quantitative type of procedure, e.g., screening, physician's office, home use, etc.;
- Summary and explanation of the test, including a short history containing methodology and the special merits and limitations of the test;
- The chemical, physical, physiological, or biological principles of the procedure.
- - The common name, if any, and quantity, proportion, or concentration or each reactive ingredient; and for biological material, the source and measure it its activity;
- - Appropriate cautions or warnings listed in 16 CFR Part 1500; the statement: "For In Vitro Diagnostic Use;" and any other limiting statements appropriate to the intended use of the product;
- - Adequate directions for reconstitution, mixing, dilution, etc.;
- - Appropriate storage instructions;
- - A statement of purification or treatment required for use; and
- - Physical, biological, or chemical indications of instability or deterioration.
- - Use or function;
- - Installation procedures and requirements;
- - Principles of operation;
- - Performance characteristics and specifications;
- - Operating instructions;
- - Calibration procedures, including equipment and/or materials;
- - Operational precautions and limitations;
- - Hazards; and
- - Service and maintenance information
- - Special precautions/preparations;
- - Additives necessary to maintain specimen integrity;
- - Known interfering substances; and
- - Recommended specimen storage, handling, and shipping instructions.
- - A list of materials provided and instruction for use, e.g., reagents, equipment, etc.;
- - A list of necessary materials that are not provided (include details such as sizes, numbers, types, and quality);
- - A description of the amounts of reagents necessary, and parameters such as time, temperature etc.;
- - A statement related to final reaction stability and any time restrictions on accurate measurements;
- - Details of calibration, identifying and listing and necessary preparation of the reference materials, samples, and blanks. Describe the calibration range including the highest and lowest values measured; and
- - Details of necessary quality control procedures and materials, e.g., positive and negative controls, acceptable performance limits.
- Explanation of the procedure for calculating the unknown, including the definition of each component of the formula, a sample calculation, and the number of significant figures appropriate for the answer;
- Limitations of the procedure, e.g., identify situations which will have an adverse impact on test results. If further testing either more specific or more sensitive, is indicated in all cases where certain results are obtained, the need for the additional test shall be stated;
- Expected values including how the range(s) was established and identify the populations on which it was established;
- Specific performance characteristics as appropriate including accuracy, specificity, precision, and sensitivity;
- * Bibliography;
- * Name and place of business of the manufacturer, packer, or distributor; and
- * Date of issuance of the last labeling revision by the firm.
Shipments or other deliveries of IVD devices are exempt from label and labeling requirements in the above headings and from standards listed under Part 861 provided the following conditions are met:
- A shipment or delivery for an investigation subject to Part 812, Investigational Device Exemption (IDE), if the device is in compliance with the subject IDE; or
- - A product in the laboratory research phase, not represented as an IVD, that is prominently labeled: "For Research Use Only. Not for use in diagnostic procedures;" and
- - A product that is being shipped or delivered for product testing prior to full commercial marketing that is prominently labeled: "For Investigational Use Only. The performance characteristics of this product have not been established."
Labeling of General Purpose Reagents and Equipment
General purpose items include routine laboratory reagents such as hydrochloric acid and equipment such as glassware whose uses are generally known by persons trained in their use. They do not need to bear the directions for use listed under Label Requirements for the Immediate Container and Labeling Requirements for Inserts and Outer Packaging, if their labeling meets the requirements listed below. If the product packaging is too small to accommodate a label with sufficient space for the labeling, and if the product is packaged in an outer container which has all of following on its labeling, then only those portions annotated with an asterisk (*) must be on the product label.
- * - A declaration of the established name, if any, and quantity, proportion, or concentration of the reagent ingredient stated in a system generally recognized by the user;
- - A statement of the purity and quality including a qualitative statement of any impurities. This can be satisfied by using a statement of conformity with a generally recognized and available standard;
- - A statement of warnings or precautions for users as contained in the regulations in 16 CFR Part 1500 and any other appropriate warnings, and the statement: "For Laboratory Use;"
- - Net quantity of contents in terms of weight or volume, or numerical count, or any combination thereof;
- * - Name and place of business of the manufacturer, packer, or distributor;
- * - A lot or control number traceable to the manufacturing history of the product; and.
- - A statement indicating the presence of and characterizing any catalytic or nonreactive ingredients e.g., buffers, preservatives, stabilizers.
- - Product labeling need include only a statement adequately describing the product, its composition, and physical characteristics if necessary for its proper use.
Other Resources
- Visit GenomeWeb on Twitter
- Visit GenomeWeb on LinkedIn
Request an Annual Quote
FDA Releases Guidance on Research Use, Investigational Use Only IVDs
NEW YORK (GenomeWeb News) – The Food and Drug Administration has released a guidance document that lays out and clarifies the rules for how in vitro diagnostic products for research use only (RUO) and investigational use only (IUO) may be used, labeled, or marketed.
FDA created the guidance on RUOs and IUOs, which has been in development for several years , because it is concerned that unapproved or uncleared IVDs are being used for clinical diagnostic use, even though their performance characteristics and manufacturing controls have not met the agency's clinical standards.
The agency allows an investigational device exemption (IDE) for medical devices that enables them to be used in research without receiving premarket approval or 510(k) clearance, but a lack of clarity in the exemption has created a loophole that makes it possible for RUOs and IOUs to seep into clinical use.
The worry is that healthcare providers could be misled about the approved applications for RUO and IUO tests, and patients could be harmed. To avoid such harms, FDA wants this guidance to inform IVD makers on how to comply and ensure that devices are being used as they were intended.
The new FDA document covers the appropriate and inappropriate uses for IVDs, as well as requirements for their labeling, manufacturing, and marketing.
To qualify as an IDE, devices must be non-invasive, must not be used as a diagnostic procedure without confirmation of the diagnosis by another medically established diagnostic, and must not require an invasive sampling procedure that presents a risk.
The guidance states that RUOs must be labeled, "For Research Use Only. Not for use in Diagnostic Procedures." IUOs must be labeled, "For Investigational Use Only. The performance characteristics of this product have not been established.''
Product labeling alone may not keep IVDs from being marketed for clinical uses, so the guidance provides details regarding how the RUOs and IUOs may be marketed, and the kinds of promotional statements that would conflict with the devices' RUO and IUO status.
Filed under
Genetic signatures halts work on molecular respiratory panel for us market, hip fracture risk linked to blood plasma protein score, top five articles on genomeweb last week: roche's nanopore sequencer, tempus ai files for ipo, more, watchmaker genomics, spt labtech partner to automate ngs library prep, bionano genomics raises $17.9m in debt financing, oxford nanopore eyes tb drug resistance for first in-house-developed diagnostic test premium, roche updates mass spec launch, instrument upgrades, upcoming tests at investor day premium, guardant health colon cancer screening test backed by majority amidst robust debate in fda meeting premium, cue health shutting down operations, tempus ai files for initial public offering.
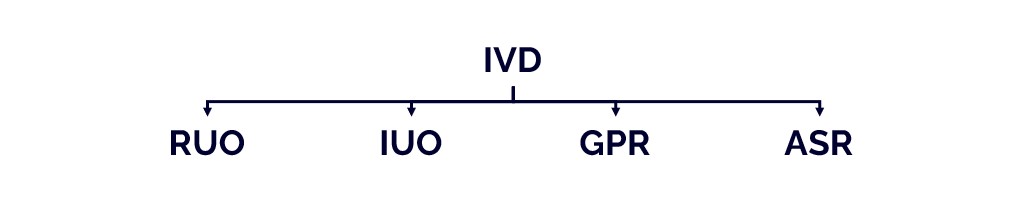
IVD Product Types: RUO, IUO, GPR, ASR
Definitions.
21 CFR Part 809.3 defines the in vitro diagnostics (IVDs) products as ”reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body’ ‘.
IVDs are devices as defined in section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), and may also be biological products subject to section 351 of the Public Health Service Act (PHS Act). Like other medical devices, IVDs are subject to premarket and postmarket controls. IVDs are generally also subject to categorization under the Clinical Laboratory Improvement Amendments (CLIA ’88) of 1988.
The U.S. regulations define four other IVD types: RUO, IUO, GPR and, ASR products.
The "Special" IVD Types

Reseach Use Only (RUO) products
RUO products are IVD products in the research laboratory phase of development. They can be under evaluation (e.g. for design evaluation, test usability) and being evaluated for developing a future commercial IVD product.
RUO products can also be used in a nonclinical laboratory context to conduct scientific research. In this case these RUO products are not intended use for further development for a clinical diagnostic use.
RUO products are not considered to be effective IVDs products (21CFR809.10(c)) and can be shipped or delivered for an investigation not subjected to an Investigational Device Exemption (IDE) as per 21CFR812.
RUO products are not subject to device classification.
RUO products shall bear the statement, prominently placed: “For Research Use Only. Not for diagnostic procedures”.
Investigational Use Only (IUO) Products
IUO products are intended be to used for product testing prior to full commercial marketing (for example, for use on specimens derived from humans to compare the usefulness of the product with other products or procedures which are in current use or recognized as useful) (21CFR809.10(c)(2)(ii).
IUO products are reagents, instruments, or test systems being used in a clinical investigation or research involving one or more subjects to determine a device’s safety and effectiveness (21CFR 812.3(h)).
IUO products are not considered to be effective IVDs products (21CFR809.10(c)) and can be shipped or delivered for an investigation not subjected to an Investigational Device Exemption (IDE) as per 21CFR812.
IUO products shall bear the statement, prominently placed: “For Investigational Use Only. The performance characteristics of this product have not been established.”
General Purpose Reagent (GPR) Products
GPR products are chemical reagents having a general laboratory application (e.g. pH buffers, isotonic solutions), used to collect, prepare, and examine specimens from human body for diagnostic purposes, and not labeled or otherwise intended for specific diagnostic application (21CFR864.4010(a)).
GPRs are medical devices that are regulated by FDA
GPR products can be individual substances or multiple substances which can be part of a diagnostic test procedure or system constituting a finished IVD test.
Most of the GPR products are classified as Class I (General Controls) and exempt from premarket notification (21CFR864.9).
GPR products shall be labeled with basic identifying information, storage conditions, warnings and precautions and shall bear the following statement: “For Laboratory Use” (21CFR809.10(d)).
Analyte Specific Reagent (ASR) Products
ASR products are “antibodies, both polyclonal and monoclonal, specific receptor proteins, ligands, nucleic acid sequences, a nd similar reagents which, through specific binding or chemical reactions with substan ces in a specimen, are intended for use in a diagnostic application for identification and quantification of an individual chemical substance or ligand in biological specim ens.” (21 CFR 864.4020(a)).
ASRs are medical devices that are regulated by FDA.
Most of the ASRs are Class I and exempt from premarket notification. These products are subject to genera l controls, including current Good Manufacturing Practices (cGMPs) (21 CFR Part 820) as well as the specific provisions of the ASR regulations (21 CFR 809.10(e), 809.30, 864.4020).
There are some ASRs that are Class II and Class III and that must be cleared or approved by FDA before they can be marketed in the United States.
An ASR is a Class II device if the reagent is used as a component in a blood banking test of a type that has been classified as a Class II device (e.g., ce rtain cytomegalovirus serological and treponema pallidum n ontreponemal test reagents) (21 CFR 864.4020(b)(2)).
An ASR is a Class III device if the reagent is intended as a component in tests intended either:
• to diagnose a contagious condition that is highly lik ely to result in a fatal outcome and prompt, accurate diagnosis offers the opportunity to mitigate the public health impact of the condition (e.g., human immunodeficiency virus (HIV/AIDS) or tuberculosis (TB)); or
• for use in donor screening for conditions for which FDA has recommended or required testing in order to safeguard th e blood supply or establish the safe use of blood and blood products (e.g., tests for hepatitis or for identifying blood groups) (21 CFR 864.4020(b)(3)). FDA considers ASRs intended to be used as a component in tests for diagnosis of HIV (including monitoring for viral load or HIV drug resistance mutations) to be Class III ASRs.
Class I ASR products shall bear the statement: “Analyte Specific Reagent. Analytical and performance characteristics are not established.”
Class II and III ASR products must bear the statement: “Analyte Specific Reagent. Except as a component of the approved/cleared test (Name of approved/cleared test), analytical and performance characteristics of this ASR are not established”.
Recommended Reading and References
Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only, Guidance for Industry and FDA Staff, November 2013
Commercially Distributed Analyte Specific Reagents (ASRs): Frequently Asked Question, Guidance for Industry and FDA Staff, September 2007
Guidance for Industry and FDA Staff Commercially Distributed Analyte Specific Reagents (ASRs): Frequently Asked Questions, September 2007
Democratizing MedTech Regulatory Knowledge
#regulatoryandmore

Template – Product Qualification and Classification under the EU IVDR 2017/746 (v.1.0)

Checklist – ISO 13485 2016 Internal Audit (v.1.0)

Template – SOP Master Validation Test Plan (v.1.0)

Template – Technical Documentation Table of Content according to the IVDR (v1.0)

Template – SOP Technical Documentation according the IVDR (v1.0)

How to Create a Project Timeline in Simple Steps

How to create a Project Timeline with VisualMaker?

The IVD Product Types: RUO, IUO, GPR, ASR

Claims under the MDR and the IVDR (Art. 7/7)

The Investigational Device Exemption (IDE)

Conformity Assessment Options for Products Failing under the MDR
Thermo Fisher Scientific
Molecular testing content for clinical lab professionals and clinicians
Paper Overview: Regulatory Guidance for Clinical Laboratories
- Experts Share Insights about Emerging HIV Drug Resistance and How to Preserve the Efficacy of Available Antiretroviral Therapies
- The Role Pharmacogenomic Testing Plays in Mental Healthcare
- Multiplex Gastrointestinal Platforms to Improve Norovirus Diagnosis and Outbreak Management

For readers outside of the U.S., this content can be used as a case study resource.
Understanding the regulatory environment is critical for the successful creation and launch of diagnostic tests—both in-vitro diagnostics (IVDs) and laboratory developed tests (LDTs). The latest educational paper published by the Arizona State University College of Health Solutions describes the regulatory landscape for laboratories establishing an LDT in the U.S., and how to navigate requirements set forth by the Food and Drug Administration (FDA) and Centers for Medicare & Medicaid Services (CMS). Let’s take a closer look at the key takeaways from this paper.
How does the FDA distinguish between IVDs and LDTs?
Laboratories that design and implement diagnostic tests must first understand how the FDA distinguishes between IVDs and LDTs (Table 1). FDA regulation of IVDs and LDTs has fueled an ongoing, two-decade debate. IVD tests manufactured for use in multiple laboratories, including clinical assays, are subject to FDA regulation such as premarket notification, approval or clearance. On the other hand, LDTs, based on in-house protocols and used at a single site, are exempt from such oversight.
Table 1. Differences between IVD tests registered with the U.S. FDA and LDTs.
*Must comply with the Clinical Laboratory Improvement Amendments (CLIA) of the U.S. Centers for Medicare and Medicaid Services.
What does the FDA say about RUOs, IUOs, and EUAs?It’s important to note that the FDA permits use of non-approved assays for research use only (RUO) or investigational use only (IUO) if they are not part of any clinical diagnostic procedures. It is illegal to make a clinical diagnosis with RUO or IUO tests because they lack comprehensive performance characterization, quality system conformity and cGMP manufacturing compliance. During a declared public health emergency, the FDA may grant Emergency Use Authorization (EUA) for unapproved medical products (including diagnostic tests). EUAs compress the development time (validation and clinical testing) required by manufacturers to provide comprehensive evidence of safety and effectiveness, compared to conventional approval. Following the EUA path is risky, and so it’s necessary to have a plan in place for market approval when the health emergency is over.How are LDTs regulated at clinical laboratories?While the FDA maintains authority over manufacturers in a device-centric manner, CMS uses a process-centric focus towards laboratories where the tests are performed. Unlike the FDA, the Clinical Laboratory Improvement Amendments (CLIA) program through CMS focuses on laboratory quality and competence, rather than the assays themselves. Any facility that performs diagnostic tests on human specimens must obtain the appropriate CLIA certificate from CMS. Analytical validation studies are required at each location to ensure assays perform as intended. CLIA-certified clinical laboratories must obtain certification for each type of assay they perform, which is assigned a complexity level by the FDA. By definition, LDTs are highly complex and must meet the most stringent criteria.Arizona State University College of Health Solutions is dedicated to translating scientific health research and discovery into practical interventions. Be sure to read ASU’s educational paper for more information on regulatory guidance for laboratories that design and implement tests for clinical use .In case you missed it, check out these related and informative resources:
- An introduction to diagnostic testing in laboratories
- Preparing for and implementing a laboratory-developed test
- Reimbursement for laboratory-developed and in vitro diagnostic tests
Intellectual property associated with laboratory-developed tests (LDTs) and in vitro diagnostic (IVD) tests
- The Simple, Sensible, Salient & Still Spell-Binding Seven Questions About Laboratory-Developed Tests
- Challenges of Establishing Laboratory-Developed Tests
- https://www.fda.gov/about-fda/what-we-do
- 21 CFR 809.3
- 42 CFR Part 493
- https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Mol Ther Methods Clin Dev
- v.24; 2022 Mar 10
Consensus guidelines for the validation of qRT-PCR assays in clinical research by the CardioRNA consortium
David de gonzalo-calvo.
1 Translational Research in Respiratory Medicine, University Hospital Arnau de Vilanova and Santa Maria, IRBLleida, 25198 Lleida, Spain
2 CIBER of Respiratory Diseases (CIBERES), Institute of Health Carlos III, 28029 Madrid, Spain
Monica Marchese
3 Integrated Bed to Bench Operations (IBBO), Luxembourg Institute of Health, 1445 Strassen, Luxembourg
Jan Hellemans
4 Biogazelle, 9052 Zwijnaarde, Belgium
5 Laboratoire National de Santé (LNS), 3555 Dudelange, Luxembourg
Nanna Lond Skov Frisk
6 Department of Science and Environment, Roskilde University, 4000 Roskilde, Denmark
7 Department of Clinical Immunology, Naestved Hospital, 4700 Naestved, Denmark
Louise Torp Dalgaard
Päivi lakkisto.
8 Minerva Foundation Institute for Medical Research, 00290 Helsinki, Finland
9 Department of Clinical Chemistry, University of Helsinki and Helsinki University Hospital, 00029 Helsinki, Finland
10 National Measurement Laboratory, LGC, Teddington TW11 0LY, UK
Andreas Scherer
11 Institute for Molecular Medicine Finland FIMM, Helsinki Institute for Life Science HiLIFE, University of Helsinki, 00014 Helsinki, Finland
12 European Research Infrastructure for Translational Medicine EATRIS, 1081 HZ Amsterdam, the Netherlands
María Laura Garcia Bermejo
13 Biomarkers and Therapeutic Targets Group, Ramon y Cajal Health Research Institute (IRYCIS), RedinRen, 28034 Madrid, Spain
Yvan Devaux
14 Cardiovascular Research Unit, Department of Population Health, Luxembourg Institute of Health, 1A-B rue Edison, 1445 Strassen, Luxembourg
Despite promising findings, quantitative PCR (qPCR)-based tests for RNA quantification have experienced serious limitations in their clinical application. The noticeable lack of technical standardization remains a huge obstacle in the translation of qPCR-based tests. The incorporation of qPCR-based tests into the clinic will benefit from guidelines for clinical research assay validation. This will ultimately impact the clinical management of the patient, including diagnosis, prognosis, prediction, monitoring of the therapeutic response, and evaluation of toxicity. However, clear assay validation protocols for biomarker investigation in clinical trials using molecular assays are currently lacking . Here, we will focus on the necessary steps, including sample acquisition, processing and storage, RNA purification, target selection, assay design, and experimental design, that need to be taken toward the appropriate validation of qRT-PCR assays in clinical research. These recommendations can fill the gap between research use only (RUO) and in vitro diagnostics (IVD). Our contribution provides a tool for basic and clinical research for the development of validated assays in the intermediate steps of biomarker research. These guidelines are based on the current understanding and consensus within the EU-CardioRNA COST Action consortium ( www.cardiorna.eu ). Their applicability encompasses all clinical areas.
Graphical abstract

These guidelines provide a tool for the development of validated assays in research. We have focused on the necessary steps that need to be taken toward the appropriate clinical research validation of qRT-PCR assays. These recommendations can fill the gap between research use only and in vitro diagnostics.
Introduction
A literature search on biomarkers and cardiovascular diseases (CVDs) highlights the poor correlation between the efforts performed in the initial steps of the development of quantitative PCR (qPCR) assay-based biomarkers, i.e., discovery and preclinical stages and their incorporation into clinical practice. There are a number of barriers that contribute to this poor implementation. The lack of technical standardization constitutes a key limitation in the incorporation of qPCR-assay-based biomarkers into the clinic. Limitations are also linked to the absence of consensus reference values, poor harmonization of the study populations, and the barriers in collaboration between academia, physicians, and industry. For instance, despite the thousands of noncoding RNA (ncRNA)-based biomarker studies published to date, there is a paucity of potential indicators that have been successfully translated into clinical practice, mainly due to the lack of reproducibility of research findings. Kok et al. 1 nicely illustrate the situation for coronary artery disease (CAD)-associated circulating microRNA (miRNA) biomarkers based on a literature review yielding 13 miRNAs found to be up- or downregulated in more than one study, of which more than half (7 out of 13) showed a contradictory result between studies (e.g., for miR-21, two studies showed upregulation and one study showed downregulation). This lack of reproducibility has also been addressed in several publications, 2 , 3 , 4 with reported causes ranging from technical analytical aspects to variable patient inclusion criteria and underpowered studies to sample quality. As such, the field of in vitro diagnostics (IVD)-grade quantitative reverse transcription PCR (qRT-PCR) assays for clinical use, initially developed in research laboratories, is still in its infancy. The incorporation of novel molecular biomarkers for clinical decision-making and patient management, i.e., diagnosis, prognosis, prediction, and monitoring of the therapeutic response or toxicity, need clear assay validation guidelines to be followed in the context of clinical research.
In this context, basic and clinical researchers often resort to the use of laboratory-developed assays with variable and undefined quality, commercial research use only (RUO) assays or, in the best-case scenarios, laboratory-developed assays validated in accordance with guidance such as minimum information for the publication of quantitative real-time PCR experiments (MIQE) guidelines. 5 The difference between such assays and certified IVD assays is significant. Laboratory-developed assays for clinical research are typically less controlled and standardized and do not need to comply with regulations, such as the European In Vitro Diagnostic Regulation (IVDR 2017/746). The European regulatory framework, based on the aforementioned IVDR and the Clinical Trials Regulation 2014/536, leaves a gray area relative to the status of laboratory assays that are used in the context of clinical trials. Poorly validated assays are not appropriate for large-scale clinical biomarker studies. Therefore, researchers would benefit from guidelines on the validation of what we refer to as clinical research (CR) assays, an assay type filling the gap between RUO and IVD that addresses the specific needs of researchers in the development of biomarkers. To some degree, such CR assays are similar to laboratory-developed test (LDT) assays in that they have undergone more thorough validation without reaching the status of a certified IVD assay.
Analytical precision (or precision): closeness of two or more measurements to each other
Analytical sensitivity: the ability of a test to detect the analyte
Analytical specificity: the ability of a test to distinguish target from nontarget analytes
Analytical trueness / analytical accuracy: closeness of a measured value to the true value
Clinical research: in this article, “clinical research” encompasses clinical studies involving patients and/or healthy controls and their biomaterials, of which the objectives are related to therapeutic interventions (clinical trials), diagnostic or prognostic developments, understanding of disease mechanisms. Good Clinical Laboratory Practice (GCLP) standards typically apply to laboratory work in clinical research
Context of use: statement that describes the appropriate use of a product or test
Fit-for-purpose: a conclusion that the level of validation associated with a medical product development tool is sufficient to support its context of use
Negative predictive value: the predictive ability of a test to identify the absence of the disease in individuals with negative test results
Positive predictive value: the predictive ability of a test to identify disease in individuals with positive results
True positive rate / Sensitivity: proportion of positives that are correctly identified
True negative rate / Specificity: proportion of negatives that are correctly identified
By defining a CR level validation, researchers can more easily license out RUO assays that are affordable and easy to obtain in the early stages of biomarker research to diagnostic test manufacturers or clinical laboratory providers. This progression is visually represented in Figure 1 . Here, we will focus on the necessary steps that need to be taken toward the appropriate validation of qRT-PCR workflows for CR and clinical use. Overall, the objective of this review is not to provide regulatory guidance for compliance with agency requirements. The aim is to provide supplementary practical and technical support in the specific context of qRT-PCR for which existing regulations are not always easy to apply or are unknown to researchers usually working outside of the regulated frameworks.

Workflow from research-use assays to in vitro diagnostic tests suitable for clinical practice
CR, clinical research; COU, context of use; IVD, in vitro diagnostics; LDT, laboratory-developed test, RUO: research use only.
Considerations for biomarker identification, validation, and clinical use
A biomarker is a characteristic, a measurable indicator of normal or pathologic biological processes, the responses to an exposure or intervention (including therapeutic interventions), or the risk of developing a medical condition or disease.
According to their intended use, biomarkers can be structured into several categories: susceptibility/risk, diagnostic, monitoring, prognostic, predictive, pharmacodynamics/response, and safety. Thus, with the right set of biomarkers, questions can be addressed such as how a condition will develop (prognostic), who will benefit from a treatment (predictive/stratification), will a treatment be efficacious (pharmacodynamic or surrogate) and beneficial (monitoring/response), will it be safe or toxic (safety), and how stable the health condition of a patient would be.
In general, the validation of a biomarker includes an evaluation of the analytical performance (trueness, precision, and analytical sensitivity and specificity) and the clinical performance (specificity, sensitivity, and predictive values). Analytical trueness (or analytical accuracy) refers to the closeness of a measured value to the true value, while analytical precision refers to the closeness of two or more measurements to each other and includes establishing the repeatability and reproducibility of the test. Analytical sensitivity is the ability of a test to detect the analyte (usually the minimum detectable concentration or LOD), and analytical specificity is the ability of a test to distinguish the target from nontarget analytes (in qPCR assays, the detection of a target sequence rather than other, nonspecific sequences).
Clinical performance is the ability of a test to correctly discriminate between the presence or absence of disease, and it is evaluated using measures of diagnostic accuracy. 6 The diagnostic sensitivity of a test is reflected in the true positive rate (TPR), meaning the correct identification of subjects with the disease, while the diagnostic specificity of a test is measured as the true negative rate (TNR), meaning the correct identification of subjects without disease. Positive predictive value (PPV) is the predictive ability of a test to identify disease in individuals with positive results, while negative predictive value (NPV) is the predictive ability of a test to identify the absence of disease in individuals with negative test results. Predictive values are dependent on the prevalence of disease.
The thresholds of these performance characteristics depend on the context of use (COU) and adhere to the “fit-for-purpose” (FFP) concept and must ideally be decided prior to the test. Properly defined, FFP is “a conclusion that the level (or rigor) of validation associated with a medical product development tool (assay) is sufficient to support its COU,” 7 where validation is the process of testing an assay performance, including the measures of the calibration of the instruments, the standardization of the experimental processes, the accuracy, the precision, and the reproducibility. 8
The COU elements are laid out in the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines and provide an informative and structured framework for identifying a biomarker's utility. 9 , 10 COU elements include (1) what aspect of the biomarker is measured and in what form, (2) what is the clinical purpose of the measurements, and (3) what is the interpretation and decision/action based on the measurements.
Biomarkers that are expected to support clinical decision-making have to be validated according to a formal qualification process, which concludes that a biomarker allows a specific interpretation and an application according to its COU in clinical product development. One example of the use of a therapeutic COU is in the allocation to specific treatment regimens. Biomarkers can influence the decision of the cessation of a patient's participation in a clinical trial, can establish a drug's proof of concept in a patient population, support clinical dose selection, and serve to enrich clinical trials for populations of interest, and can help evaluate treatment responses. This concept can be generalized to all phases of discovery research and drug development with the term FFP.
Hence, the intended use of a biomarker determines the choice of analytical methods as well as the stringency of the performance criteria of the biomarker during its validation process. An adjustment of methods with regard to new findings during the FFP validation process may be necessary and may concern the selection of preanalytical conditions. A validation phase with preliminary performance acceptance criteria, followed by larger sample sizes and off-site tests of the biomarker assay, is essential for the determination of the robustness and usability in more extended settings.
Nonclinical biomarkers (RUOs), which are the output of the FFP validation process, do not require regulatory submission. RUO biomarkers can be used as “good-enough” biomarkers internally or possibly for publication. However, full validation will be required in case the biomarker will be developed further toward clinical use, e.g., as a companion diagnostic.
For regulatory acceptance, a biomarker needs to follow and fully comply with the path of a qualification that outlines how a biomarker will be used in the clinical setting. The outcome will be a valid biomarker within its COU, one that is measured in an analytical test system with well-established performance characteristics and for which there is an established framework or body of evidence that elucidates the physiological, toxicological, pharmacological, or clinical significance of the test results. 9 , 10 , 11
Controlled experimental environments, which include experimental designs with predefined acceptance criteria, preanalytical requirements, qualified equipment, trained operators, analytical performance, and data stewardship, are prerequisites for reliable and thereby meaningful measurement of biomarkers.
Guidelines for validation of CR-grade qPCR assays
Many references on validation requirements exist, ranging from academic guidelines and international standards such as ICH and ISO to national regulatory requirements such as the 21 CFR Part 820 (US) and IVDR 2017/746 (EU). 8 , 12 , 13 A practical challenge for the use of many of these standards is the fact that they have not been tailored to the specifics of PCR-based tests. ISO 20395:2019 (Biotechnology — Requirements for evaluating the performance of quantification methods for nucleic acid target sequences — qPCR and digital PCR [dPCR]), which was recently published, addresses specific underpinning requirements. Inspired by these standards, we present guidelines for the practical analytical validation of CR-grade qPCR assays in clinical samples.
It is worth noting that the concept of a guideline explicitly allows researchers to deviate from them as a function of the specific needs and characteristics of their tests. A few examples where a risk analysis could alter the validation of qPCR assays include the following: (1) the specificity of an assay is typically a crucial assay property (for reference genes, this requirement may be relaxed since their measurements are required to represent the total amount of RNA, not necessarily the amount of a specific gene); (2) the limit of detection (LOD) is an important property for assays testing weakly expressed genes or for tests optimized to work with minimal input quantities (for tests where the analyte is highly abundant, defining the LOD addresses specific points in standards and regulatory requirements without significantly contributing to the quality of the assay); or (3) the capacity of an assay to accurately quantify small expression differences requires a thorough validation when used for biomarkers that rely on such small expression differences. On the other hand, less extensive validation experiments may be sufficient for tests that rely on the detection of large expression differences.
Method development for RUO
Even when aiming for a higher level of analytical assay validation, it is useful to start with RUO-level validation. Such validation is fast and easy and allows for the elimination of inferior designs before proceeding to the more extensive CR (or ultimately IVD)-level validation.
Prior to the initiation of the analytical validation, the qPCR master mix components and the thermocycler and cycling conditions must be selected, optimized, and then fixed. Frequently, these decisions are implicit by relying on standard procedures used within the laboratory, but one needs to be aware that the obtained validation results only apply to that setting. As there are considerable differences between qPCR master mixes with regard to assay performance, polymerase reliability, and optimized conditions, defining the conditions is crucial. Importantly, validated results only apply to the settings used, meaning that revalidation is required whenever the setup changes.
RUO-level analytical validation (for genes of interest and reference genes) should at least cover the following aspects: (1) correct amplification on a positive control sample (for example, Cq values below 28), (2) lack of amplification on a negative control sample, (3) evidence of RT and amplification efficiency, and (4) specificity of an amplified product (melting temperature and/or amplicon length analysis). An experimental workflow providing data for validation could be performed as follows:
- i. In general, RNA, rather than cDNA, is preferred as a positive control. The RNA may be derived from a positive sample or generated by in vitro transcription from a vector or synthetic sequence.
- ii. cDNA sample. Ideally derived from an RNA sample that has undergone the same process (collection, extraction, RT) as clinical samples and that is known to express the gene of interest. Alternatively, this could also be a commercial sample available in large quantities and with sufficient and similar RNA quality to the intended test samples. Clinical biobanks, under ISO certification, may be a source of samples if no commercial controls are available.
- iii. A cloned and sequence-verified DNA fragment containing the transcript of interest.
- iv. A synthetic DNA fragment containing the transcript sequence of interest.
- i. No-template control testing for contamination and primer-dimer formation (e.g., water or carrier RNA).
- ii. Genomic DNA testing for gDNA coamplification. A RT reaction without reverse transcriptase added will also test for gDNA contamination of the RNA eluate.
- iii. “Extraction blank” to control for contamination at the extraction stage.
- iv. Optional: cDNA from other species to test for cross-species reactivity (important in preclinical studies where human genes are used in a hybrid context).
- v. Optional: cDNA from a specific RNA sample known not to contain the transcript or variant of interest (this is the ideal negative control because of its complex matrix similar to real samples).
- ◦ cDNA, if the gene of interest is highly expressed.
- ◦ Plasmid or long double-stranded synthetic DNA fragments.
- ◦ Short single-stranded chemically synthesized templates, e.g., 60-mer oligonucleotides of the first and last 30 nucleotides of the amplicon. This solution can be applied also for genes with very low or rare expression. Of note, for probe-based assays, the oligonucleotide (oligo) should also contain the probe binding site.
- ◦ In-vitro -transcribed synthetic RNA fragments representing the entire amplicon.
- ◦ RNA from cell lines, patient samples, and/or reference materials.

Considerations for sample material for qPCR dilution series
The last option is applicable to one-step qRT-PCR.
- ii. Six-point (or more) 10-fold dilution series, ideally spanning the 10 1 to 10 6 copies/qPCR reaction range. If the input quantities for this dilution series are well known (because of the use of qualified reference samples or because of calibration by means of dPCR), the dilution series could also be used for absolute quantification.
- • In total, 21–33 reactions/assay.
- • Perform triplicate qPCR reactions for all samples.
- • Amplicon size analysis should be performed (agarose gel or microfluidic electrophoresis). Positive control samples should yield a single sized product of correct length. For negative controls with a signal, it may support troubleshooting by differentiating between primer-dimer formation and template contamination.
- • Evaluate amplification plots for correct amplification in positive control samples.
- i. Evaluate melting curves.
- ii. Positive control samples should yield a characteristic Tm peak. Different tools enable the prediction of the Tm peak (or multiple peaks for some amplicons with nonuniform GC% distributions) as a reference value.
- iii. Negative samples should display no primer-dimer peak (typically broader peak at low Tm) nor a template Tm peak.
- • Evaluate negative control samples. Ideally, no amplification is observed. In cases where the sequence of interest is very highly expressed, a low level of false positive signals (high Cq value) might be tolerated.
- i. Linearity in a standard curve. Deviations from linearity may be observed at the ends of the dilution series, e.g., because of plateauing due to contamination or primer-dimer formation or because of inhibition due to the use of too much cDNA. The testable range is restricted to the linear range of the dilution series.
- ii. Amplification efficiency. Ideally in the 90%–110% range. Efficiencies of 80%–90% or 110%–120% are suboptimal but acceptable for some assays, e.g., when the effect size (difference) is sufficiently large and such small technical imperfections do not interfere with their separation.
CR-level validation
A CR assay consists of the entire workflow from RNA template measurement to data analysis. The standardized treatment of samples at each step is imperative to ensure biomarker performance.
Reverse transcription
During reverse transcription, the complementary DNA template is synthesized. Ideally, this reaction should generate a 1-to-1 DNA complementary to the RNA template, but this is very rarely achievable. Depending on the type and quality of RNA investigated, a reverse transcription priming method should be selected. The RT reaction may use random primers (6–9 mers), oligo dT primers, a combination of both of the aforementioned primers, or target primers, corresponding to those used in the qPCR reaction. Validation experiments should include the test of saturation by an RNA template, which is performed as multiple cDNA synthesis reactions with increasing amounts of purified RNA, including an RNA spike-in (an external control) followed by qPCR for the intended targets. Here, there should be linearity between the Cq levels of the targets and the input amount of RNA within a range corresponding to (patho)physiological levels of the RNA template. Alternatively, increasing amounts of synthetic target RNA can be added to multiple reverse transcription reactions of one specific RNA sample. Similarly, to test for reaction inhibition during cDNA synthesis, we also recommend adding a synthetic spike-in RNA molecule during reverse transcription that is different from the one used for spike-in during RNA isolation. The amount of RNA spike-in should generally be in the linear dynamic range of the assay, which would often be in the attomol range of added spike-in. This is fundamental to correct for differences in the RT reaction efficiency.
Target selection
Different approaches lead to the identification of target sequences. Target selection is beyond the scope of these guidelines but depends on the following questions: is one interested in the gene with all its isoforms or in a subset of transcripts encoding a particular protein? Does the assay have to detect a specific allele or fusion event? Are there any regions to be avoided for assay design because of homology with mouse sequences that would interfere with analysis of murine xenografts?
Reference genes
Importantly, most qPCR tests measuring gene expression levels rely on reference genes for data normalization. Assay validation is thus not limited to the assays measuring the target of interest but must include assays for selected reference genes as well. Since the quality of the final test results strongly depends on the quality of normalization by means of the selected reference genes, it is important to ensure that multiple and stably expressed reference genes are selected. A pilot study evaluating eight candidate reference genes in 12 representative samples and analyzed by a tool such as geNorm 14 or NormFinder 15 can provide the needed assurance that the reference genes are unaffected by the experimental conditions and are stable in the population and sample type of interest. Additionally, for small RNAs determined in body fluids, a set of reference small RNAs has already been suggested. 16 Nevertheless, a critical evaluation of these reference small RNAs is fundamental for each experimental condition and study population.
Assay design
To increase the success rate of analytical assay validation, the basics of a design by tools such as Primer3 17 should be complemented with additional bioinformatic analyses testing for the specificity, transcript coverage, and absence of secondary structures and common single-nucleotide polymorphisms (SNPs) in primer or probe annealing regions. Such analyses may be integrated in assay design by programs such as primerXL, 18 performed independently with online tools such as BiSearch (for specificity) 19 and UNAfold (secondary structures), 20 or performed manually in genome browsers (transcript coverage and SNP overlap).
Despite the benefit of such in silico examinations, they cannot provide a full guarantee of optimal performance when put in practice in the lab. Therefore, multiple designs can be fed into downstream analytical validation, allowing for the selection of the best performing assay and increasing the likelihood of identifying at least one assay meeting all requirements.
Experimental design for CR assay validation of qPCR-based tests
We provide an example of a design that may be used for most gene expression tests ( Figure 3 ). This design may, however, have to be modified to meet the requirements of certain specific analytical COUs, e.g., when dealing with transcript or allele-specific assays, when working in a multispecies context, or when aiming for multiplexing. The proposed number of biological replicates may also be increased, e.g., for biomarkers with a small effect size.

Clinical research qPCR assay development
The researcher also needs to predefine the assay performance acceptance criteria. These might relate to parts of the method (for instance, the qPCR amplification efficiency) but should always include criteria for the full workflow starting from the matrix to the end result. The latter are typical test-related criteria such as accuracy, precision, specificity, sensitivity, LOD, and limit of quantification (LOQ). The results of this validation have to be properly documented, e.g., electronically.
Due to the lack of suitable reference samples, the accuracy of normalized gene expression levels is difficult to determine. Inspired by the mixing experiments proposed in the microarray quality control (MAQC) studies but enhanced to ensure the testability of differential expression for any gene, we propose an approach relying on known mixtures of in-vitro -transcribed RNAs to support the accuracy analysis of qRT-PCR tests. To make this experiment representative of the actual analysis applying reference gene normalization, it includes both transcripts for the gene(s) of interest (GOI) and for the reference gene(s).
To systematically assess the robustness of the test for RNA integrity, we propose the evaluation of a series of RNA samples with varying degrees of artificial degradation. This degradation can be achieved via heating, sonication, UV radiation, or incubation with ribonucleases. 21 , 22 If samples are known to systematically yield high-quality RNA, and its integrity is systematically tested, one may consider skipping this robustness analysis. Evaluation of the quality of RNA can be achieved by spectrophotometry and by microfluidic electrophoretic methods based on the RNA integrity number (RIN) or an equivalent metric. It is more difficult to evaluate the quality of ncRNA, such as miRNAs, but this can be done either by verifying the presence of ubiquitous ncRNA molecules (e.g., RNU-24, miR-16, miR-221) or by measuring quality scores that have been described in specific COUs. 23 , 24
Consensus experimental design for a standard gene expression assay is presented hereafter (for absolute and relative quantification). The recommended acceptance criteria for precision and accuracy depends on the intended purpose of the assay:
- i. GOI template containing amplicon sequences for the GOIs: promotor – GOI1 – GOI2 – … – GOIn – control. If a large number of assays for different GOIs are to be tested, one may consider making different GOI templates, with each containing a subset of GOI amplicon sequences.
- ii. REF template containing amplicon sequences for the reference genes: promotor – REF1 – REF2 – … – REFn – control.
- • Optional: determine the copy number of the synthetic templates by means of dPCR using the control assay. If dPCR is not available, one has to rely on the quantities described by the oligo producer to estimate the template copy number.
- • Create a dilution series for both GOI and REF templates spanning the 5 × 10 6 to 5 copies/PCR reaction range.
- • Confirm the correctness of the dilution series by testing the control assay. With qPCR, the correctness of fold dilutions can be verified. If dPCR is available, the accuracy of the dilution points can also be assessed. If these deviate more than 2-fold (or any fold difference that is relevant for the intended purpose), a new dilution series should be made.
- • Heat-treat an RNA sample for different times (75°C for 1–10 min/μg RNA). Ideally, the same representative RNA sample (same matrix and extraction method) should be used. If such RNA is already too degraded to be used as a starting point for the RNA integrity series or the available material is too limiting for these experiments, one may have to rely on commercial samples such as the MAQC RNA.
- • Assess RNA integrity by means of microfluidic electrophoresis (see above). The process of artificial RNA degradation and integrity assessment may have to be iterated until proper timings have been found to generate the targeted RNA integrity range. Create cDNA for the selected RNA samples.
- • For DNA analysis, an equivalent test can be set up.
- • In vitro transcription of the two templates containing the GOI and REF, respectively (two separate reactions).
- i. A: mixture containing equal amounts of GOI and REF RNA.
- ii. B: mixture containing the same amount of REF RNA as sample A but only 1/1,000 of GOI RNA.
- i. C: 90% A + 10% B
- ii. D: 75% A + 25% B
- iii. E: 50% A + 50% B
- iv. F: 25% A + 75% B
- v. G: 10% A + 90% B
- • Spike mock RNA with synthetic RNA mixes A–G. The mock RNA provides for a more natural, complex RNA background. Any RNA sample void of amplifiable sequences may be suitable. RNA from bacteriophage MS2 is often a good candidate mock RNA sample.
- • Synthesize cDNA (by performing RT) for all spiked RNA samples and for unspiked mock RNA (sample H).
- • For gDNA analyses, an equivalent test, not needing in vitro transcription and cDNA synthesis, can be set up.
- • Four representative clinical samples with sufficient material should be selected for at least 2 extractions. If material for individual samples is limiting, one may consider homogeneous pooling and mixing of samples to obtain samples with sufficient material to support repeat extraction.
- • Perform repeat extraction, reflecting the different sources of variation (day of extraction, extraction kit lot number, person executing the extraction).
- • Perform cDNA synthesis independently for the two sets of 4 RNA extracts.
- • In total, 48–96 reactions/assay.
- i. Dilution series of synthetic DNA templates (see Preparation 1).
- ii. No-template control (NTC) with carrier RNA.
- iii. 5 cDNA samples from RNA integrity series.
- iv. 3 cDNA samples from representative clinical samples.
- • Assess specificity on amplicons from the 3 representative cDNA samples using size and, for assays using intercalating dyes, melt curve analysis.
- i. Determine slope, intercept, linear dynamic range, coefficient of variation (r 2 ), and amplification efficiency from dilution series data.
- ii. Determine qPCR repeatability for cDNA from representative clinical samples and for the different samples of the dilution series. The latter may reveal concentration-dependent repeatability, typically showing increasing variability as concentrations approach the detection limit.
- iii. Assessment of the absence or degree of primer-dimer formation on the NTC sample.
- iv. Assess the robustness of the assay against RNA degradation. A robust assay would show identical normalized relative quantity (NRQ) values for artificially degraded RNA as for intact, undegraded RNA. Lower NRQ values for degraded samples would reveal an impact of certain degrees of RNA degradation.
- • In total, 48 reactions/assay.
- i. On 12 samples: an NTC, the 7 cDNA samples (A–G) derived from the RNA/cDNA titration series (see Preparation 3), and 4 cDNA samples derived from the same repeat extraction and reverse transcription.
- ii. Using assays for both the reference genes and a set of GOIs.
- iii. Triplicate qPCR reactions (or other replicate number as will be used in later testing—more replicates may improve data quality).
- iv. Repeat the analysis of the 12 samples twice within the same run. One set containing the 4 cDNA samples of one extraction and reverse transcription round, and the other set containing those of repeat extraction and reverse transcription.
- i. Calculate normalized expression levels for the GOIs by normalizing their relative quantities with the geometric mean of the relative quantities of the selected reference genes. 25
Relative input quantities
- iii. Determine repeatability by comparing the normalized expression levels for the two repeats within the same run.
- i. Prepared by a different person.
- ii. On a different day.
- iii. Measured on a different qPCR instrument.
- • Determine between-run precision of the assay by comparing the NRQ values for the two repeats between the runs. The results for samples A–G only reflect the repeatability of the qPCR measurements, including pipetting errors. The results for the other 4 samples also reflect the variability of other parts of the workflow (RNA extraction and cDNA synthesis).
- 8 .Validation of a run.
Quality control steps to monitor relative quantification assay performance and accept or reject a run include the following: (1) cDNA prepared from a reference RNA sample should show Cq < 30; (2) reverse transcription NTC sample should give indeterminate Cq or Cq at least 5 Cq units higher than the highest Cq value of the positive test samples used in the assay; (3) NTC sample should give indeterminate Cq or Cq at least 5 Cq units higher than the highest Cq value of the positive test samples used in the assay; (4) if amplification efficiencies are remeasured, they should fall within their predefined acceptance range (typically 90%–110%); or (5) if multiple reference genes are used to normalize data, their stability measures should fall within their predefined acceptance range (typically geNorm M value < 0.5). 14 , 25
Quality control steps to monitor absolute quantification assay performance and accept or reject a run include the following: (1) NTC sample gives an undetermined result or a result below the lowest point of the calibration curve, (2) the calibration curve R 2 >99%, (3) the calibration curve slope is between −2.9 and −3.8, or (4) there are at least four consecutive data points in the calibration curve (defining the “validated range” for the run).
The guidelines proposed above, which are summarized in Figure 4 , are not mandatory and are proposed to assist researchers in validating their assays before implementation in CR and clinical trials. They are not intended to impose formal validation requirements but rather to describe the consensus obtained within the European CardioRNA consortium (CardioRNA COST Action CA17129) on a relevant set of validation experiments. These guidelines are focused on the analytical validation of singleplex gene expression assays. Several aspects are explicitly not covered: multiplex assays, data analysis, cross-species amplification (xenograft, infectious), preamplification, genotyping or allele-specific PCRs, or dPCR. These assays may require other approaches that are inspired by these guidelines or are completely tailor made.

Checklist for reference procedure
We envisage reviewing these guidelines in 3 years based on the feedback received from the CR community, the practical experience gained in using these guidelines, the new consensus in the scientific community, and new technological developments and approaches.
Conclusions, remarks, and perspectives
The increase in RNA-focused research in the last decade has led to great advances in the general and specific knowledge of the transcriptome and pathophysiological mechanisms of diseases, leading to the identification of new biomarkers that are useful in clinical practice. Despite the promising findings, qPCR-based tests for RNA quantification have experienced serious limitations in their direct clinical application. The development and commercialization of novel qRT-PCR-based tools is a laborious process, and successful assay validation requires substantial resources. Ultimately, the establishment and application of evidence-based recommendations for CR assays may reduce the time and cost of obtaining new assays from the research laboratory to clinical practice and the market.
These recommendations can fill the gap between RUO and IVD. They are the output of a collective effort of the EU-CardioRNA consortium with collaboration and endorsement by the European Research Infrastructure for Translational Medicine (EATRIS) ( https://www.EATRIS.eu ) and the Biobanking and BioMolecular Resources Research Infrastructure (BBMRI) ( www.BBMRI.eu ), with both being part of the EU-AMRI alliance (European Alliance of Medical Research Infrastructures), whose vision is to alleviate the attrition rate of biomarkers early in their development to assure accuracy and to help save costs, time, and expectancies of patients and clinicians. We are confident that application of these guidelines will result in more effective biomarkers development for many diseases but that are, above all, useful in clinical practice.
Acknowledgments
This manuscript is based upon work from EU-CardioRNA COST Action CA17129 ( www.cardiorna.eu ) supported by COST (European Cooperation in Science and Technology). D.d.G.-C. wants particularly to acknowledge Marta Molinero and Jo Vandesompele for their technical support. D.d.G.-C. has received financial support from the Instituto de Salud Carlos III (Miguel Servet 2020: CP20/00041), which is co-funded by the European Social Fund (ESF)/"Investing in your future.” This work is supported by the Instituto de Salud Carlos III (PI20/00577), which is co-funded by European Regional Development Fund (ERDF)/“A way to make Europe.” CIBERES is an initiative of the Instituto de Salud Carlos III. Y.D. is funded by the EU Horizon 2020 project COVIRNA (grant agreement 101016072), the National Research Fund (grants C14/BM/8225223, C17/BM/11613033, and COVID-19/2020-1/14719577/miRCOVID), the Ministry of Higher Education and Research, and the Heart Foundation-Daniel Wagner of Luxembourg. P.L. is funded by the Finnish Cultural Foundation, The Finnish Foundation for Cardiovascular Research, The Finnish Society of Clinical Chemistry, and the Finnish Foundation for Laboratory Medicine.
Author contributions
D.d.G.-C., M.M., J.H., F.B., N.L.S.F., A.S., L.T.D., and P.L. designed the article and wrote the manuscript. C.F., M.L.G.B., and Y.D. revised the manuscript. All authors read and approved the final manuscript.
Declaration of interests
D.d.G.-C. holds a patent on miRNAs as biomarkers. Y.D. holds patents related to diagnostic and therapeutic applications of RNAs. The other authors declare no competing interests.
Research Use Only or IVD: What’s Right for Your Lab?
by Tina Sobania | Clinical , Molecular

Publish Date: September 13, 2018
There are many misconceptions in the clinical industry regarding laboratory quality control materials. With numerous products available and manufacturers using various labeling practices, how do you know what’s best for your laboratory?
To help clear up the confusion, we’re answering two important questions clinical laboratorians have about quality control products.
Are diagnostic system controls IVDs?
One common misconception is that materials used for quality control of diagnostic systems are not themselves in vitro diagnostics (IVDs). However, the U.S. Food & Drug Administration (FDA) has written regulations citing quality control material as medical devices. For example, 21 CFR 862.1660 , Mulit-Analyte Controls Unassayed under Clinical Chemistry, and more recently 21 CFR 866.3920 , classify Class II controls requiring FDA 510(k) review under microbiology.
It’s important to understand that if a manufacturer for controls of nucleic acid amplification states its product works with a specific instrument or assay in its labeling or marketing literature, the FDA considers the material to be a Class II IVD and requires a 510(k) review . The FDA has established special controls for this type of material to ensure the product is properly labeled, performs according to claims and remains stable. In addition, IVD material must be manufactured under the FDA’s current Good Manufacturing Practices (cGMP).
Should “Research Use Only” products be used for quality control?
The second misconception clinical laboratories should be aware of involves material labeled as Research Use Only (RUO). RUO labeling is intended for products that are still under development and are not commercially distributed. A developer would use this labeling to ship product for “investigation relating to product development” as explained by the FDA in guidance document, Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigation Use Only .
Another factor one must consider is products labeled RUO are not required to be manufactured in accordance with cGMP and FDA Quality System Regulation. Lack of manufacturing controls may be detrimental to the quality of the control material. As such, clinical laboratories using RUO quality control materials to ensure the quality of testing may be placing patients at unnecessary risk.
Key Takeaway
To maintain the highest possible quality of your diagnostic testing, it’s best to choose materials that have been manufactured by a cGMP compliant facility under the FDA QSR, and when necessary reviewed by the FDA. Materials clearly labeled as IVDs provide that assurance and lower your laboratory’s risk.
Follow the links below to find all the FDA regulations cited in this post.
- 21 CFR 862.1660 CFR – Code of Federal Regulations Title 21, Subchapter H – Medical Devices
- 21 CFR 866.3920 CFR – Code of Federal Regulations Title 21, Subchapter H – Medical Devices
- Distribution of In Vitro Diagnostic Products Labeled for Research Use Only of Investigation Use Only

Written by Tina Sobania
You may also like.

MLS and MLT Career Opportunities
As a manufacturer of quality controls used in clinical diagnostics, we see firsthand the difficulties that laboratory...

The Challenges of Diagnosing and Treating Secondary Infections
In the past two decades, there have been six major global outbreaks of infectious diseases. While these infections may...
Trackbacks/Pingbacks
- Put Your Best Plate Forward – #CreepyCultures is Back! – Microbiologics Blog - […] Read Next – Research Use Only or IVD: What’s Right for Your Lab? […]
- Clinical Case File: Group A Streptococcus – Microbiologics Blog - […] Read Next – Research Use Only or IVD: What’s Right for Your Lab? […]
- Dear Stanley: Molecular QC Best Practices – Microbiologics Blog - […] Read Next – Research Use Only or IVD: What’s Right for Your Lab? […]
- Our Top Posts from 2018 – Microbiologics Blog - […] 1. Research Use Only or IVD: What’s Right for Your Lab? […]
- LDT Oversight Deliberation Continues – Microbiologics Blog - […] Read Next – Research Use Only or IVD: What’s Right for Your Lab? […]

- Pharmaceutical
- Events and Webinars
- Uncategorized
1-800-599-2847 microbiologics.com [email protected]
QUICK LINKS
CATEGORIES RESOURCES ABOUT US CONTACT US SITE MAP PRIVACY POLICY

research use only Diagnostics
Research use only diagnostics.
Staff at Catachem have been pioneers in the early development of diagnostic reagents for use on automated equipment going back to the 1970’s, and the company continues to develop research versions of new reagents for future use in the human clinical laboratory.
Catachem’s diagnostic products have been evolving for the nearly 40 years that the company has been in business. Landmark papers by Catachem’s founder Luis P. Leon, on a UV glucose reagent have been cited in the scientific literature 43 times at a recent count. Some newer testing products in Catachem’s U.S. research portfolio include plasma free hemoglobin (PFH), ethylene glycol, methanol, and glutamate dehydrogenase all of which can be used on most the modern laboratory analyzers.

Specialty Testing Products for Research Use
Plasma free hemoglobin (c462-0a), ethylene glycol – quantitative test (c504-0a).
Catachem has developed an accurate quantitative test (C504-0A) that determines ethylene glycol levels. Values obtained on Catachem’s quantitative test reagent compare closely to values obtained on GC/MS methods (Am.J.Clin.Pathol. 2011, 136:165-6). Catachem’s product has been designed to effectively eliminate interferences from propylene glycol, a secondary component in some antifreeze products that is also used as an additive in some human foodstuffs.
NOTE: The antidote 4-methylpyrazole (fomepizole), shows no interference to a level of 120mg/L. A typical target range for treatment is 8 – 25mg/L.
(Annals of Clinical Biochemistry, Robson et al, 09, 2016)
Methanol – Quantitative Test (C604-0A)
Ethylene glycol – fastox test kit (c504-0c), methanol – fastox test kit (c604-fq).
Designed similarly to Catachem’s FasTox ethylene glycol test for the low-volume user, this 340 nm test for methanol is for use on a simple spectrophotometer. Three individual tests are packaged along with three low and three high controls, eliminating the need for additional calibration. This product has an 18-month shelf life.
*For research use only
The above products for research use only in the USA
Applies to all products
How Can We Help You?
If you are looking to place an order, request technical help or ask questions about us, get in touch today!
VISIT US AT ADLM IN CHICAGO! JULY 30TH – AUGUST 1ST, 2024 BOOTH #4844
Language selection
- Français fr
Important notice
Temporary website updates are in progress to fix technical issues. For assistance, visit our Contact Us page.
The Sidney Laboratory

CFIA science laboratory brochure: The Sidney Laboratory (PDF – 2,400 kb)
- Renewing the Sidney Centre for Plant Health
The Government of Canada is investing in the renewal of the Canadian Food Inspection Agency's (CFIA's) Sidney Laboratory (Centre for Plant Health) on Vancouver Island.
The Sidney Laboratory is located on the traditional territory of the W̱SÁNEĆ peoples, which include the W̱JOŁEŁP (Tsartlip), the W̱SĺḴEM (Tseycum),the SȾÁUTW̱ (Tsawout), the BOḰEĆEN (Pauquachin) and the MÁLEXEȽ (Malahat) First Nations.
About the Canadian Food Inspection Agency
The Canadian Food Inspection Agency (CFIA) is a science-based regulator with a mandate to safeguard the food supply, protect the health of plants and animals , and support market access. The Agency relies on high-quality, timely and relevant science as the basis of its program design and regulatory decision-making. Scientific activities inform the Agency's understanding of risks, provide evidence for developing mitigation measures, and confirm the effectiveness of these measures.
CFIA scientific activities include laboratory testing, research, surveillance, test method development, risk assessments and expert scientific advice. Agency scientists maintain strong partnerships with universities, industry, and federal, provincial and international counterparts to effectively carry out the CFIA's mandate.

The Sidney Laboratory, also known as the Centre for Plant Health, is Canada's only post-entry quarantine (PEQ), research and diagnostic facility for virus testing of all fruit-bearing trees, grapevines and small fruit (e.g. berries). PEQ facilities ensure the safe introduction of foreign plant material into Canada.
A renewal project at the Sidney Laboratory will build a new world-class plant health diagnostic and research facility that will provide CFIA scientists and partners with state-of-the-art amenities to advance plant science. Having the right tools is essential to help develop and partner on new ideas and opportunities to protect Canada's plant resources and to grow the agriculture and agri-food sector.
- Pathogen testing of imported tree fruits, small fruits and grapevine (i.e. bacteria, virus and virus-like organisms).
- Export certification for the trade of tree fruits, small fruits, and grapevine.
- Elimination of virus infections from valuable fruit and grape varieties.
- Maintenance of the national repository of Generation 1, virus-tested tree fruit and grapevine varieties for Canadian export certification and domestic distribution.
- Support trade through participation on international panels to develop harmonized standards for the movement and testing of plant materials.
Plant-related research
- Develop and validate methods for supporting quarantine and virus testing activities.
- Validate and apply new technologies such as Next Generation Sequencing.
- Identify and characterize new viruses and virus-like diseases.
- Develop rapid, sensitive, molecular diagnostic tests to support the implementation of the Plant Protection Act .
Scientific techniques
Enzyme-linked immunosorbent assay (elisa).
- ELISA determines the presence of a particular substance (e.g. food allergens, toxins, or pathogens) using antibodies that bind to specific target protein(s). A subsequent reaction producing a detectable signal such as colour change shows the presence of the target substance. The strength of the signal gives an indication of the amount present in the sample.
Polymerase Chain Reaction assay (PCR)
- PCR is a technique that can detect a pathogen in a plant sample by targeting nucleic acid (e.g. DNA or RNA) that is specific to the pathogen of interest. It then amplifies the target until it is detectable. The presence of the amplified nucleic acid indicates the presence of the pathogen in the original sample.
Woody-host bioassays
- Bioassay of a woody-host (i.e. plant that produces wood as its structural tissue) starts with an indicator plant that is disease-free and is then inoculated with a sample from a plant with an unknown health status. The indicator plant is then monitored for any symptoms of disease. If symptoms are observed, then the original plant that was used for the inoculation harbours a pathogen.
Herbaceous-host bioassays
- Bioassay of an herbaceous (i.e. non- woody) disease-free indicator plant is inoculated with sap from a plant with an unknown health status. The indicator plant is then monitored for any symptoms of disease. If symptoms are observed, then the original plant that was used for the inoculation harbours a pathogen.
Quality management
All CFIA laboratories are accredited in accordance with the International Standard ISO/IEC 17025, General requirements for the competence of testing and calibration laboratories . The Standards Council of Canada (SCC) provides accreditation for routine testing, test method development and non-routine testing, as identified on the laboratory's Scope of Accreditation on the SCC website. Accreditation formally verifies the CFIA's competence to produce accurate and reliable results. The results are supported by the development, validation and implementation of scientific methods, conducted by highly qualified personnel, using reliable products, services, and equipment, in a quality controlled environment. Participation in international proficiency testing programs further demonstrates that our testing is comparable to laboratories across Canada and around the world.
Physical address
Sidney Laboratory (Centre for Plant Health) 8801 East Saanich Road North Saanich, British Columbia V8L 1H3
More information
- Digging through the past and planting seeds for the future
- News release: Celebrating a site blessing and ground breaking at the Centre for Plant Health
- Meet Tracy Lawrence, CFIA virology technician
- Podcast with Anna-Mary Schmidt, Head of Grapevine Diagnostics
- News release: Government of Canada invests $80 million in the Centre for Plant Health
Learn about other CFIA laboratories .
- Introduction
- Conclusions
- Article Information
Data Sharing Statement
See More About
Sign up for emails based on your interests, select your interests.
Customize your JAMA Network experience by selecting one or more topics from the list below.
- Academic Medicine
- Acid Base, Electrolytes, Fluids
- Allergy and Clinical Immunology
- American Indian or Alaska Natives
- Anesthesiology
- Anticoagulation
- Art and Images in Psychiatry
- Artificial Intelligence
- Assisted Reproduction
- Bleeding and Transfusion
- Caring for the Critically Ill Patient
- Challenges in Clinical Electrocardiography
- Climate and Health
- Climate Change
- Clinical Challenge
- Clinical Decision Support
- Clinical Implications of Basic Neuroscience
- Clinical Pharmacy and Pharmacology
- Complementary and Alternative Medicine
- Consensus Statements
- Coronavirus (COVID-19)
- Critical Care Medicine
- Cultural Competency
- Dental Medicine
- Dermatology
- Diabetes and Endocrinology
- Diagnostic Test Interpretation
- Drug Development
- Electronic Health Records
- Emergency Medicine
- End of Life, Hospice, Palliative Care
- Environmental Health
- Equity, Diversity, and Inclusion
- Facial Plastic Surgery
- Gastroenterology and Hepatology
- Genetics and Genomics
- Genomics and Precision Health
- Global Health
- Guide to Statistics and Methods
- Hair Disorders
- Health Care Delivery Models
- Health Care Economics, Insurance, Payment
- Health Care Quality
- Health Care Reform
- Health Care Safety
- Health Care Workforce
- Health Disparities
- Health Inequities
- Health Policy
- Health Systems Science
- History of Medicine
- Hypertension
- Images in Neurology
- Implementation Science
- Infectious Diseases
- Innovations in Health Care Delivery
- JAMA Infographic
- Law and Medicine
- Leading Change
- Less is More
- LGBTQIA Medicine
- Lifestyle Behaviors
- Medical Coding
- Medical Devices and Equipment
- Medical Education
- Medical Education and Training
- Medical Journals and Publishing
- Mobile Health and Telemedicine
- Narrative Medicine
- Neuroscience and Psychiatry
- Notable Notes
- Nutrition, Obesity, Exercise
- Obstetrics and Gynecology
- Occupational Health
- Ophthalmology
- Orthopedics
- Otolaryngology
- Pain Medicine
- Palliative Care
- Pathology and Laboratory Medicine
- Patient Care
- Patient Information
- Performance Improvement
- Performance Measures
- Perioperative Care and Consultation
- Pharmacoeconomics
- Pharmacoepidemiology
- Pharmacogenetics
- Pharmacy and Clinical Pharmacology
- Physical Medicine and Rehabilitation
- Physical Therapy
- Physician Leadership
- Population Health
- Primary Care
- Professional Well-being
- Professionalism
- Psychiatry and Behavioral Health
- Public Health
- Pulmonary Medicine
- Regulatory Agencies
- Reproductive Health
- Research, Methods, Statistics
- Resuscitation
- Rheumatology
- Risk Management
- Scientific Discovery and the Future of Medicine
- Shared Decision Making and Communication
- Sleep Medicine
- Sports Medicine
- Stem Cell Transplantation
- Substance Use and Addiction Medicine
- Surgical Innovation
- Surgical Pearls
- Teachable Moment
- Technology and Finance
- The Art of JAMA
- The Arts and Medicine
- The Rational Clinical Examination
- Tobacco and e-Cigarettes
- Translational Medicine
- Trauma and Injury
- Treatment Adherence
- Ultrasonography
- Users' Guide to the Medical Literature
- Vaccination
- Venous Thromboembolism
- Veterans Health
- Women's Health
- Workflow and Process
- Wound Care, Infection, Healing
Get the latest research based on your areas of interest.
Others also liked.
- Download PDF
- X Facebook More LinkedIn
Zhong A , Amat MJ , Anderson TS, et al. Completion of Recommended Tests and Referrals in Telehealth vs In-Person Visits. JAMA Netw Open. 2023;6(11):e2343417. doi:10.1001/jamanetworkopen.2023.43417
Manage citations:
© 2024
- Permissions
Completion of Recommended Tests and Referrals in Telehealth vs In-Person Visits
- 1 Center for Primary Care, Harvard Medical School, Boston, Massachusetts
- 2 Division of General Medicine and Primary Care, Beth Israel Deaconess Medical Center, Boston, Massachusetts
- 3 Division of General Medicine, Brigham and Women’s Hospital, Boston, Massachusetts
- 4 Healthcare Systems Engineering Institute, Northeastern University, Boston, Massachusetts
- 5 Stanford University School of Medicine, Stanford, California
Question Is there a difference in the rate of completion for diagnostic tests and referrals ordered during a telehealth visit compared with those ordered during an in-person visit?
Findings In this cohort study of 4133 diagnostic tests and referrals (colonoscopies, cardiac stress tests, and dermatology referrals) ordered between March 1, 2020, and December 31, 2021, at 2 affiliated clinical primary care sites, 58% of those ordered during in-person visits were completed within the designated time frame compared with 43% of those ordered during telehealth visits. The rate of completion was between 40% and 65% for all test types, regardless of visit modality.
Meaning The findings of this study suggest that rates of completion for diagnostic tests and referrals were low for all visit types but worse when ordered during telehealth visits.
Importance Use of telehealth has increased substantially in recent years. However, little is known about whether the likelihood of completing recommended tests and specialty referrals—termed diagnostic loop closure —is associated with visit modality.
Objectives To examine the prevalence of diagnostic loop closure for tests and referrals ordered at telehealth visits vs in-person visits and identify associated factors.
Design, Setting, and Participants In a retrospective cohort study, all patient visits from March 1, 2020, to December 31, 2021, at 1 large urban hospital-based primary care practice and 1 affiliated community health center in Boston, Massachusetts, were evaluated.
Main Measures Prevalence of diagnostic loop closure for (1) colonoscopy referrals (screening and diagnostic), (2) dermatology referrals for suspicious skin lesions, and (3) cardiac stress tests.
Results The study included test and referral orders for 4133 patients (mean [SD] age, 59.3 [11.7] years; 2163 [52.3%] women; 203 [4.9%] Asian, 1146 [27.7%] Black, 2362 [57.1%] White, and 422 [10.2%] unknown or other race). A total of 1151 of the 4133 orders (27.8%) were placed during a telehealth visit. Of the telehealth orders, 42.6% were completed within the designated time frame vs 58.4% of those ordered during in-person visits and 57.4% of those ordered without a visit. In an adjusted analysis, patients with telehealth visits were less likely to close the loop for all test types compared with those with in-person visits (odds ratio, 0.55; 95% CI, 0.47-0.64).
Conclusions The findings of this study suggest that rates of loop closure were low for all test types across all visit modalities but worse for telehealth. Failure to close diagnostic loops presents a patient safety challenge in primary care that may be of particular concern during telehealth encounters.
The use of telehealth has increased substantially in recent years, by more than 60-fold in 2020. 1 Although telehealth made up less than 1% of medical visits before the COVID-19 pandemic, it became ubiquitous with the onset of the pandemic, before tapering to still-unforeseen levels, with 37% of adults reporting at least 1 telephone or video visit in 2022. 2 Past studies have examined patient experiences with telehealth, finding a high degree of satisfaction due to the ease of use, improved communication, and reduced wait and travel times. 3 - 6 Studies have also found high levels of diagnostic concordance (defined as degree of agreement) overall between telehealth and in-person visits. 7 , 8 However, little is known about how telehealth affects a patient’s likelihood of completing tests and specialty referrals—termed diagnostic loop closure . 9
Failure to close referral loops is one of the leading causes of diagnostic errors in primary care. This is of particular importance with telehealth, given the finding of reduced diagnostic concordance 10 in primary care due to the breadth of potential diagnoses. 7 , 9 The eventual outcomes of these oversights can be catastrophic; an estimated 12 million diagnostic errors occur annually in the US, contributing to 64 000 preventable deaths and costing the health care system $100 billion per year. 11 , 12
In this study, we examined the prevalence of the failure to close diagnostic loops for 3 representative types of tests and referrals ordered during primary care telehealth visits compared with those ordered during in-person visits and those ordered without a visit. We hypothesized that orders placed during telehealth visits were less likely to be completed due to a variety of patient, clinician, and system-level factors, which we sought to explore further.
We conducted a retrospective cohort study at 2 primary care sites in Boston, Massachusetts: 1 large urban hospital-based primary care practice and 1 affiliated community health center. We examined all visits from March 1, 2020, to December 31, 2021. The hospital-based site cares for approximately 42 000 patients while the affiliated community health center cares for approximately 6000 patients. We accessed clinical data for the 2 sites through a data warehouse, which maintains data for both and allows search by billing diagnoses, tests, referrals, and patient demographic characteristics. At both sites, patients have the option of both in-person visit and telehealth visits (telephone or video). Our research protocol was approved by the institutional review boards at Beth Israel Deaconess Medical Center and Harvard Medical School. The institutional review boards at both institutions deemed that informed consent from study participants was not required because only deidentified data were involved. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology ( STROBE ) reporting guideline. 13
We reviewed referrals and tests resulting from a concerning finding or need for screening, prompting the clinician to order an additional diagnostic workup. The order may have been placed during an in-person visit, a telehealth visit, or on a date when no visit occurred. We extracted data for 3 types of high-risk tests and referrals ordered between March 1, 2020, and December 31, 2021: colonoscopy referrals, dermatology referrals for suspicious skin lesions, and cardiac stress tests (including both exercise and chemical stress tests, with and without imaging). These tests were chosen due to their different characteristics, ease of test completion, high-risk nature with major clinical implications, and institutional prioritization. We then examined all orders for completion between March 1, 2020, to March 30, 2022.
Within all visit modalities, the order for testing or referrals is placed electronically through the electronic medical record. There is little to no proactive outreach for the scheduling of patients for routine orders. During an in-person visit, the patient is given a form with a telephone number to call to schedule the test or referral. For patients with limited English proficiency or complex needs, a member of the support staff team may help the patient schedule this test at checkout. During a telehealth visit, a patient is given the telephone number to call to schedule the test or referral by the clinician during the visit. In all scenarios, the patient receives no communication after the visit reminding them to schedule the test or referral.
Our main independent variable was the modality of the patient visit during which the diagnostic test or referral was ordered (in-person, telehealth [telephone and video], or ordered without a visit). Our primary outcome was failure to close the loop on the test or referral. The loop was considered closed if the orders were completed within 365 days for colonoscopy, 90 days for dermatology, and 45 days for cardiac stress test referrals. Time intervals for loop closure were determined through review of the literature, in conversations with physicians administering the tests based on the urgency of making a diagnosis, and with the goal of ensuring adequate time for patients to schedule despite current systematic delays.
We included several patient factors obtained from the electronic medical record as independent covariates. These included demographic variables such as age, sex assigned at birth (female or male), self-reported race (Asian, Black, White, or unknown or other), self-reported ethnicity (Hispanic, non-Hispanic, or unknown), educational level (high school or less, college, or unknown), primary language (English, other, or unknown), and insurance type (Medicaid, Medicare, commercial, or unknown or other), which we regarded as a surrogate for income level. Race and ethnicity were included due to the importance of identifying disparities in care as a first step toward addressing them. Race was divided into the following categories: American Indian or Alaska Native, Asian, Black or African American, White, declined to answer, other race or mixed race, prefer not to say, unable to obtain, and unknown or not specified. For ease of reporting, it was narrowed down ( Table 1 ). The unknown or other category includes American Indian or Alaska Native, declined to answer, other race or mixed race, prefer not to say, unable to obtain, and unknown or not specified.
We also included patients’ chronic illness burden and psychiatric illness burden as covariates. Chronic illness burden was calculated using the Charlson comorbidity index (0, 1-2, or ≥3). 14 This was determined using the billed diagnosis codes for all medical visits within 2 years of the index referral date. Psychiatric illness burden was based on a billing code for depression ( International Statistical Classification of Diseases and Related Health Problems, Tenth Revision code F32) at a visit before or on the index referral date, using the date range (yes or no). Other markers of psychiatric illness, such as anxiety, were not examined.
Other covariates described the clinical site (hospital-based primary care practice and affiliated community health center), type of clinician ordering the test or referral (attending physician, resident physician, or nurse practitioner), and whether the patient had ever used the patient portal to review a visit note, which we used as a surrogate for patient engagement. We additionally included a marker for pause of elective procedures at our institution to represent periods of high COVID-19 infectivity, which occurred from March 13 to June 9, 2020; December 7, 2020, to March 1, 2021; and November 21, 2021, to January 27, 2022.
We compared patient characteristics between the 3 groups: patients with orders placed during in-person visits, telehealth visits, and no visit. We then performed a univariate analysis with loop closure as the outcome. For the primary comparison of loop closures associated with telehealth visits with in-person visits, we adjusted for all the patient factors listed above using multivariable logistic regression. Additionally, we performed a stepwise regression analysis to identify the extent to which groups of covariates contributed to the findings (ie, we sequentially adjusted for grouped covariates to determine which group best explained the univariate result). The stepwise regression analysis first adjusted for the test type, then patient demographic characteristics and comorbidities, then clinical site and clinician type, and finally patient engagement. For tests of statistical significance, analysis of variance was used for continuous variables and χ 2 analysis was used for all other variables. We conducted all statistical analyses using Stata, version 16.1 (StataCorp LLC), with statistical significance set at a 2-sided α value of .05.
Among 4133 patients with test and referral orders, the mean (SD) age was 59.3 (11.7) years; 2163 (52.3%) were women and 1970 (47.7%) were men; 203 (4.9%) were Asian, 1146 (27.7%) were Black, 2362 (57.1%) were White, and 422 (10.2%) had an unknown or other race ( Table 1 ). The 4133 orders included colonoscopy referrals (n = 3254), dermatology referrals for suspicious skin lesions (n = 455), and cardiac stress tests (n = 424). Of these, 2167 (52.4%) were ordered during an in-person visit, 1151 (27.8%) were ordered during a telehealth visit, and 815 (19.7%) were ordered without a visit. Compared with patients with in-person visits, patients with telehealth visits were more likely to be White, have higher educational attainment, have English as their primary language, have commercial insurance, and to have been seen by an attending physician ( Table 1 ).
Of the 1151 orders placed during a telehealth visit, 490 (42.6%) were completed within the designated time frame vs 58.4% (1265 of 2167) placed during an in-person visit and 57.4% (468 of 815) placed without a visit ( Figure 1 ). In the univariate analysis, patients with telehealth visits were less likely to close the loop on high-risk tests and referrals overall compared with in-person visits (odds ratio [OR], 0.53; 95% CI, 0.46-0.61) ( Table 2 ). Patients with orders placed without an in-person or telehealth visit were equally likely as patients with an in-person visit to close the loop on ordered tests and referrals. A stepwise multivariable regression that adjusted for groups of covariates sequentially did not result in significant changes in rates of loop closure when comparing telehealth visits with in-person visits ( Table 3 ). In the adjusted analysis, patients with telehealth visits were still less likely to close the loop on all tests and referral types overall (OR, 0.55; 95% CI, 0.47-0.64). When adjusting for periods of high COVID-19 infectivity, the overall odds of loop closure were 0.58 (95% CI, 0.51-0.67) across all referrals and test orders. The rate of loop closure plateaued before the allotted time frame expired for all test types (colonoscopies, 365 days; dermatology referrals, 90 days; cardiac stress tests, 45 days) and the time to loop closure did not appear to differ between orders originating from telehealth vs in-person visits ( Figure 2 ).
We identified a total of 3254 colonoscopy referrals, which made up 78.7% of all orders; 998 (30.7%) were ordered during a telehealth visit, 1570 (48.2%) were ordered during an in-person visit, and 686 (21.1%) were ordered without a visit. Of the colonoscopy referrals ordered at a telehealth visit, 39.8% (n = 397) were completed within 365 days, compared with 56.9% (n = 893) ordered at an in-person visit and 56.7% (n = 389) ordered without a visit. Before adjustment for the covariates, patients with telehealth visits were less likely to close the loop on colonoscopy referrals compared with in-person visits (OR, 0.50; 95% CI, 0.43-0.59). This remained consistent in the adjusted analysis (OR, 0.49; 95% CI, 0.42-0.58) ( Table 2 ).
We identified a total of 455 dermatology referrals for suspicious skin lesions, which made up 11.0% of all orders; 65 (14.3%) were ordered during a telehealth visit, 301 (66.2%) were ordered during an in-person visit, and 89 (19.6%) were ordered without a visit. Of the dermatology referrals ordered at a telehealth visit, 63.1% (n = 41) were completed within 90 days, compared with 61.5% (n = 185) ordered at an in-person visit and 62.9% (n = 56) ordered without a visit. There was no significant difference between telehealth and in-person visits for dermatology referral loop closure in the unadjusted (OR, 1.07; 95% CI, 0.61-1.87) or adjusted (OR, 1.07; 95% CI, 0.60-1.95) analyses ( Table 2 ).
We identified a total of 424 cardiac stress tests, which made up 10.3% of all orders; 88 (20.8%) were ordered during a telehealth visit, 296 (69.8%) were ordered during an in-person visit, and 40 (9.4%) were ordered without a visit. Of the stress tests ordered at a telehealth visit, 59.1% (n = 52) were completed within 45 days, compared with 63.2% (n = 187) ordered at an in-person visit and 57.5% (n = 23) ordered without a visit. There was no significant difference between telehealth and in-person visits for stress test loop closure in the unadjusted (OR, 0.84; 95% CI, 0.52-1.37) or adjusted (OR, 0.84; 95% CI, 0.49-1.43) analyses ( Table 2 ).
Loop closure rates for selected diagnostic tests and specialty referrals were unacceptably low across all visit modalities. Furthermore, in a multivariable analysis, patients with telehealth visits were substantially less likely to close open loops compared with patients with in-person visits. In subgroup analyses based on test type, patients with telehealth visits were less likely to close the loop on colonoscopies, but statistically significant differences were not detected for urgent dermatology referrals or stress test orders. Sequentially adjusting for test type, patient demographic characteristics, comorbidities, clinical site, clinician type, and patient engagement did not markedly change the odds of loop closure in the stepwise regression analysis.
As reported, the rate of loop closure was low (between 40% and 65%) for all test types, regardless of visit modality, compared with an acceptable threshold of 80% in population health metrics and an ideal goal of 95% in models based on expert consensus. 15 This is consistent with research that has found suboptimal completion rates for tests ordered in urgent care scenarios and suggests that common system factors may play a more important role in loop closure than visit modality. 16 Thus, while the differences in loop closure between telehealth and in-person visits may be concerning, system-level changes are needed to improve test completion rates across all modalities. These might include automated tracking for outstanding tests within electronic medical records and interventions such as telephone outreach to patients, automated text and email reminders, and the use of referral managers. 17 These considerations may be particularly important for patients who rely heavily on telehealth, such as those in remote rural areas and disadvantaged patients with limited health access and literacy.
When investigating notable differences in loop closure for orders placed during telehealth visits, our findings suggest that differences in loop closure may be inherent to telehealth as a modality. One potential mechanism to explain this may be the lack of systems in place to help patients complete test and referral orders. During in-person visits, members of the support staff team sometimes help patients schedule their tests at checkout; however, this support is absent during telehealth visits. After the visit, patients do not receive any communication reminding them to schedule the test or referral, which may further limit loop closure. Other potential explanations include the possibility that it may be more difficult to remember information provided during telehealth visits, that telehealth may present unique communication barriers, or that it may be more difficult to engage patients in shared decision-making during virtual visits, thus decreasing patient engagement with test and referral orders. 18 - 21
These barriers may be more pronounced for colonoscopies compared with other types of orders given the increased difficulty in preparing for and completing the invasive procedure, thus explaining the disparities in loop closure compared with the other orders included in our analysis. 22 Furthermore, given the notable difference in loop closure for colonoscopies, telehealth visits may serve as a marker for patients not participating in a high-complexity test. They may also highlight an important association between test indication and test completion, with screening indication yielding lower priority for both the patient and physician. This builds on past work that has identified lower rates of patient adherence to hemoglobin A 1c testing recommendations during telehealth encounters. 23 With this association in mind, an order for a routine test placed during a telehealth visit might serve as a process marker to trigger additional assistance or alternative ways to complete the indicated test. Our observation that telehealth does not seem to worsen loop closure for tests prompted by patient symptoms or concerns (suspicious skin lesions or chest pain) suggests that patient activation may be a key factor in closing loops and may overcome some of the challenges inherent to telehealth visits.
The widespread failure to close diagnostic test and referral loops identified in this study may also provide a mechanism to explain past findings that primary care practices with higher telehealth use are associated with increased rates of acute care visits and that primary care telehealth visits are associated with increased emergency department encounters, 24 , 25 possibly due, at least in part, to a failure to close the loop on ordered tests. These findings should be contextualized within the literature, which has identified significant advantages to telehealth vis-à-vis convenience, access, and diagnostic concordance. 5 - 8 Nonetheless, our findings highlight a potential limitation to telehealth that, to our knowledge, had not been previously identified.
Strengths of this study include the novelty of the inquiry, variety of tests evaluated, adjustment for confounding, and patient characteristics that are consistent with prior reports evaluating the composition of telehealth patient populations. 1 , 2 This study has limitations. First, the study lacks data on the urgency of orders, scheduled tests and referrals that were canceled, and clinical outcomes resulting from the failure to close diagnostic loops. The CIs for estimates of loop closures when comparing telehealth visits with in-person visits for dermatology referrals and stress tests were wide, suggesting that our smaller sample sizes may have reduced our opportunity to identify important differences. Similarly, although the study used data from 2 different primary care settings, there were too few visits at the community site to detect significant differences. We lack data to fully explain why orders without a visit had completion rates that were similar to those of face-to-face visits. Whether the orders resulted from a patient telephone call or message was not available in the data set. In addition, the multivariable stepwise analysis suggests possible issues with residual confounding.
In this cohort study, completion rates for diagnostic tests and referrals were low for all treatment modalities at 2 primary care sites, but worse for telehealth. These results are among the first to document how telehealth affects patients’ likelihood of completing recommended tests and specialty referrals. Telehealth visits present novel opportunities to reduce diagnostic errors and may play an increasingly important role in clinical practice given the continued expansion of telehealth following the COVID-19 pandemic.
Accepted for Publication: October 3, 2023.
Published: November 15, 2023. doi:10.1001/jamanetworkopen.2023.43417
Open Access: This is an open access article distributed under the terms of the CC-BY License . © 2023 Zhong A et al. JAMA Network Open .
Corresponding Author: Maelys J. Amat, MD, MBA, Beth Israel Deaconess Medical Center, 330 Brookline Ave, Boston, MA 02215 ( [email protected] ).
Author Contributions: Dr Amat had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Mr Zhong and Dr Amat shared first authorship.
Concept and design: Zhong, Amat, Anderson, Sternberg, Fernandez, Schiff, Benneyan, Phillips.
Acquisition, analysis, or interpretation of data: Anderson, Shafiq, Sternberg, Salant, Fernandez, Schiff, Aronson, Benneyan, Singer, Phillips.
Drafting of the manuscript: Zhong, Amat, Phillips.
Critical review of the manuscript for important intellectual content: Anderson, Shafiq, Sternberg, Salant, Fernandez, Schiff, Aronson, Benneyan, Singer.
Statistical analysis: Anderson, Shafiq, Fernandez, Benneyan.
Obtained funding: Schiff, Benneyan, Singer, Phillips.
Administrative, technical, or material support: Amat, Sternberg, Schiff, Aronson, Benneyan.
Supervision: Amat, Fernandez, Schiff, Phillips.
Conflict of Interest Disclosures: Dr Anderson reported receiving grants from the American Heart Association, US Deprescribing Research Network, and American College of Cardiology outside the submitted work. Dr Phillips reported holding small equity interest in Bicycle Health and Grow Therapy outside the submitted work, serving as an expert witness for Morrison Mahoney LLP, and being an unpaid member of the scientific advisory board of Maven Project, a nonprofit organization. No other disclosures were reported.
Funding/Support: This work was funded by Agency for Healthcare Research and Quality (AHRQ) grant R18 5R18HS027282. Dr Amat was supported by the Arnold Tofias and Leo Condakes Quality Scholarship Program. Dr Anderson was supported by a grant from the National Institute on Aging (K76AG074878).
Role of the Funder/Sponsor: The funding organizations had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Disclaimer: The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the AHRQ or the National Institutes of Health.
Data Sharing Statement: See the Supplement 1 .
Additional Contributions: Dru Ricci, BA (Harvard Medical School Center for Primary Care), assisted with the preparation of the manuscript. No financial compensation was provided.
- Register for email alerts with links to free full-text articles
- Access PDFs of free articles
- Manage your interests
- Save searches and receive search alerts
Suggestions or feedback?
MIT News | Massachusetts Institute of Technology
- Machine learning
- Social justice
- Black holes
- Classes and programs
Departments
- Aeronautics and Astronautics
- Brain and Cognitive Sciences
- Architecture
- Political Science
- Mechanical Engineering
Centers, Labs, & Programs
- Abdul Latif Jameel Poverty Action Lab (J-PAL)
- Picower Institute for Learning and Memory
- Lincoln Laboratory
- School of Architecture + Planning
- School of Engineering
- School of Humanities, Arts, and Social Sciences
- Sloan School of Management
- School of Science
- MIT Schwarzman College of Computing
Adhesive coatings can prevent scarring around medical implants
Press contact :, media download.

*Terms of Use:
Images for download on the MIT News office website are made available to non-commercial entities, press and the general public under a Creative Commons Attribution Non-Commercial No Derivatives license . You may not alter the images provided, other than to crop them to size. A credit line must be used when reproducing images; if one is not provided below, credit the images to "MIT."

Previous image Next image
When medical devices such as pacemakers are implanted in the body, they usually provoke an immune response that leads to buildup of scar tissue around the implant. This scarring, known as fibrosis, can interfere with the devices’ function and may require them to be removed.
In an advance that could prevent that kind of device failure, MIT engineers have found a simple and general way to eliminate fibrosis by coating devices with a hydrogel adhesive. This adhesive binds the devices to tissue and prevents the immune system from attacking it.
“The dream of many research groups and companies is to implant something into the body that over the long term the body will not see, and the device can provide therapeutic or diagnostic functionality. Now we have such an ‘invisibility cloak,’ and this is very general: There’s no need for a drug, no need for a special polymer,” says Xuanhe Zhao, an MIT professor of mechanical engineering and of civil and environmental engineering.
The adhesive that the researchers used in this study is made from cross-linked polymers called hydrogels, and is similar to a surgical tape they previously developed to help seal internal wounds. Other types of hydrogel adhesives can also protect against fibrosis, the researchers found, and they believe this approach could be used for not only pacemakers but also sensors or devices that deliver drugs or therapeutic cells.
Zhao and Hyunwoo Yuk SM ’16, PhD ’21, a former MIT research scientist who is now the chief technology officer at SanaHeal, are the senior authors of the study, which appears today in Nature . MIT postdoc Jingjing Wu is the lead author of the paper.
Preventing fibrosis
In recent years, Zhao’s lab has developed adhesives for a variety of medical applications, including double-sided and single-sided tapes that could be used to heal surgical incisions or internal injuries. These adhesives work by rapidly absorbing water from wet tissues, using polyacrylic acid, an absorbent material used in diapers. Once the water is cleared, chemical groups called NHS esters embedded in the polyacrylic acid form strong bonds with proteins at the tissue surface. This process takes about five seconds.
Several years ago, Zhao and Yuk began exploring whether this kind of adhesive could also help keep medical implants in place and prevent fibrosis from occurring.
To test this idea, Wu coated polyurethane devices with their adhesive and implanted them on the abdominal wall, colon, stomach, lung, or heart of rats. Weeks later, they removed the device and found that there was no visible scar tissue. Additional tests with other animal models showed the same thing: Wherever the adhesive-coated devices were implanted, fibrosis did not occur, for up to three months.
“This work really has identified a very general strategy, not only for one animal model, one organ, or one application,” Wu says. “Across all of these animal models, we have consistent, reproducible results without any observable fibrotic capsule.”
Using bulk RNA sequencing and fluorescent imaging, the researchers analyzed the animals’ immune response and found that when devices with adhesive coatings were first implanted, immune cells such as neutrophils began to infiltrate the area. However, the attacks quickly quenched out before any scar tissue could form.
“For the adhered devices, there is an acute inflammatory response because it is a foreign material,” Yuk says. “However, very quickly that inflammatory response decayed, and then from that point you do not have this fibrosis formation.”
One application for this adhesive could be coatings for epicardial pacemakers — devices that are placed on the heart to help control the heart rate. The wires that contact the heart often become fibrotic, but the MIT team found that when they implanted adhesive-coated wires in rats, they remained functional for at least three months, with no scar tissue formation.
“The formation of fibrotic tissue at the interface between implanted medical devices and the target tissue is a longstanding problem that routinely causes failure of the device. The demonstration that robust adhesion between the device and the tissue obviates fibrotic tissue formation is an important observation that has many potential applications in the medical device space,” says David Mooney, a professor of bioengineering at Harvard University, who was not involved in the study.
Mechanical cues
The researchers also tested a hydrogel adhesive that includes chitosan, a naturally occurring polysaccharide, and found that this adhesive also eliminated fibrosis in animal studies. However, two commercially available tissue adhesives that they tested did not show this antifibrotic effect because the commercially available adhesives eventually detached from the tissue and allowed the immune system to attack.
In another experiment, the researchers coated implants in hydrogel adhesives but then soaked them in a solution that removed the polymers’ adhesive properties, while keeping their overall chemical structure the same. After being implanted in the body, where they were held in place by sutures, fibrotic scarring occurred. This suggests that there is something about the mechanical interaction between the adhesive and the tissue that prevents the immune system from attacking, the researchers say.
“Previous research in immunology has been focused on chemistry and biochemistry, but mechanics and physics may play equivalent roles, and we should pay attention to those mechanical and physical cues in immunological responses,” says Zhao, who now plans to further investigate how those mechanical cues affect the immune system.
Yuk, Zhao, and others have started a company called SanaHeal, which is now working on further developing tissue adhesives for medical applications.
“As a team, we are interested in reporting this to the community and sparking speculation and imagination as to where this can go,” Yuk says. “There are so many scenarios in which people want to interface with foreign or manmade material in the body, like implantable devices, drug depots, or cell depots.”
The research was funded by the National Institutes of Health and the National Science Foundation.
Share this news article on:
Related links.
- Xuanhe Zhao
- Hyunwoo Yuk
- Jingjing Wu
- Department of Mechanical Engineering
- Department of Civil and Environmental Engineering
Related Topics
- Mechanical engineering
- Civil and environmental engineering
- Biomedical engineering
- Medical devices
- National Institutes of Health (NIH)
- National Science Foundation (NSF)
Related Articles

Engineers develop surgical “duct tape” as an alternative to sutures

Double-sided tape for tissues could replace surgical sutures

Super-strong surgical tape detaches on demand

Paper-folding art inspires better bandages
Previous item Next item
More MIT News

Controlled diffusion model can change material properties in images
Read full story →

Sophia Chen: It’s our duty to make the world better through empathy, patience, and respect

Using art and science to depict the MIT family from 1861 to the present

Convening for cultural change

Q&A: The power of tiny gardens and their role in addressing climate change

In international relations, it’s the message, not the medium
- More news on MIT News homepage →
Massachusetts Institute of Technology 77 Massachusetts Avenue, Cambridge, MA, USA
- Map (opens in new window)
- Events (opens in new window)
- People (opens in new window)
- Careers (opens in new window)
- Accessibility
- Social Media Hub
- MIT on Facebook
- MIT on YouTube
- MIT on Instagram
- SUGGESTED TOPICS
- The Magazine
- Newsletters
- Managing Yourself
- Managing Teams
- Work-life Balance
- The Big Idea
- Data & Visuals
- Reading Lists
- Case Selections
- HBR Learning
- Topic Feeds
- Account Settings
- Email Preferences
Research: What Companies Don’t Know About How Workers Use AI
- Jeremie Brecheisen

Three Gallup studies shed light on when and why AI is being used at work — and how employees and customers really feel about it.
Leaders who are exploring how AI might fit into their business operations must not only navigate a vast and ever-changing landscape of tools, but they must also facilitate a significant cultural shift within their organizations. But research shows that leaders do not fully understand their employees’ use of, and readiness for, AI. In addition, a significant number of Americans do not trust business’ use of AI. This article offers three recommendations for leaders to find the right balance of control and trust around AI, including measuring how their employees currently use AI, cultivating trust by empowering managers, and adopting a purpose-led AI strategy that is driven by the company’s purpose instead of a rules-heavy strategy that is driven by fear.
If you’re a leader who wants to shift your workforce toward using AI, you need to do more than manage the implementation of new technologies. You need to initiate a profound cultural shift. At the heart of this cultural shift is trust. Whether the use case for AI is brief and experimental or sweeping and significant, a level of trust must exist between leaders and employees for the initiative to have any hope of success.
- Jeremie Brecheisen is a partner and managing director of The Gallup CHRO Roundtable.
Partner Center
A .gov website belongs to an official government organization in the United States.
A lock ( ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
- About Adverse Childhood Experiences
- Risk and Protective Factors
- Program: Essentials for Childhood: Preventing Adverse Childhood Experiences through Data to Action
- Adverse childhood experiences can have long-term impacts on health, opportunity and well-being.
- Adverse childhood experiences are common and some groups experience them more than others.

What are adverse childhood experiences?
Adverse childhood experiences, or ACEs, are potentially traumatic events that occur in childhood (0-17 years). Examples include: 1
- Experiencing violence, abuse, or neglect.
- Witnessing violence in the home or community.
- Having a family member attempt or die by suicide.
Also included are aspects of the child’s environment that can undermine their sense of safety, stability, and bonding. Examples can include growing up in a household with: 1
- Substance use problems.
- Mental health problems.
- Instability due to parental separation.
- Instability due to household members being in jail or prison.
The examples above are not a complete list of adverse experiences. Many other traumatic experiences could impact health and well-being. This can include not having enough food to eat, experiencing homelessness or unstable housing, or experiencing discrimination. 2 3 4 5 6
Quick facts and stats
ACEs are common. About 64% of adults in the United States reported they had experienced at least one type of ACE before age 18. Nearly one in six (17.3%) adults reported they had experienced four or more types of ACEs. 7
Preventing ACEs could potentially reduce many health conditions. Estimates show up to 1.9 million heart disease cases and 21 million depression cases potentially could have been avoided by preventing ACEs. 1
Some people are at greater risk of experiencing one or more ACEs than others. While all children are at risk of ACEs, numerous studies show inequities in such experiences. These inequalities are linked to the historical, social, and economic environments in which some families live. 5 6 ACEs were highest among females, non-Hispanic American Indian or Alaska Native adults, and adults who are unemployed or unable to work. 7
ACEs are costly. ACEs-related health consequences cost an estimated economic burden of $748 billion annually in Bermuda, Canada, and the United States. 8
ACEs can have lasting effects on health and well-being in childhood and life opportunities well into adulthood. 9 Life opportunities include things like education and job potential. These experiences can increase the risks of injury, sexually transmitted infections, and involvement in sex trafficking. They can also increase risks for maternal and child health problems including teen pregnancy, pregnancy complications, and fetal death. Also included are a range of chronic diseases and leading causes of death, such as cancer, diabetes, heart disease, and suicide. 1 10 11 12 13 14 15 16 17
ACEs and associated social determinants of health, such as living in under-resourced or racially segregated neighborhoods, can cause toxic stress. Toxic stress, or extended or prolonged stress, from ACEs can negatively affect children’s brain development, immune systems, and stress-response systems. These changes can affect children’s attention, decision-making, and learning. 18
Children growing up with toxic stress may have difficulty forming healthy and stable relationships. They may also have unstable work histories as adults and struggle with finances, jobs, and depression throughout life. 18 These effects can also be passed on to their own children. 19 20 21 Some children may face further exposure to toxic stress from historical and ongoing traumas. These historical and ongoing traumas refer to experiences of racial discrimination or the impacts of poverty resulting from limited educational and economic opportunities. 1 6
Adverse childhood experiences can be prevented. Certain factors may increase or decrease the risk of experiencing adverse childhood experiences.
Preventing adverse childhood experiences requires understanding and addressing the factors that put people at risk for or protect them from violence.
Creating safe, stable, nurturing relationships and environments for all children can prevent ACEs and help all children reach their full potential. We all have a role to play.
- Merrick MT, Ford DC, Ports KA, et al. Vital Signs: Estimated Proportion of Adult Health Problems Attributable to Adverse Childhood Experiences and Implications for Prevention — 25 States, 2015–2017. MMWR Morb Mortal Wkly Rep 2019;68:999-1005. DOI: http://dx.doi.org/10.15585/mmwr.mm6844e1 .
- Cain KS, Meyer SC, Cummer E, Patel KK, Casacchia NJ, Montez K, Palakshappa D, Brown CL. Association of Food Insecurity with Mental Health Outcomes in Parents and Children. Science Direct. 2022; 22:7; 1105-1114. DOI: https://doi.org/10.1016/j.acap.2022.04.010 .
- Smith-Grant J, Kilmer G, Brener N, Robin L, Underwood M. Risk Behaviors and Experiences Among Youth Experiencing Homelessness—Youth Risk Behavior Survey, 23 U.S. States and 11 Local School Districts. Journal of Community Health. 2022; 47: 324-333.
- Experiencing discrimination: Early Childhood Adversity, Toxic Stress, and the Impacts of Racism on the Foundations of Health | Annual Review of Public Health https://doi.org/10.1146/annurev-publhealth-090419-101940 .
- Sedlak A, Mettenburg J, Basena M, et al. Fourth national incidence study of child abuse and neglect (NIS-4): Report to Congress. Executive Summary. Washington, DC: U.S. Department of Health an Human Services, Administration for Children and Families.; 2010.
- Font S, Maguire-Jack K. Pathways from childhood abuse and other adversities to adult health risks: The role of adult socioeconomic conditions. Child Abuse Negl. 2016;51:390-399.
- Swedo EA, Aslam MV, Dahlberg LL, et al. Prevalence of Adverse Childhood Experiences Among U.S. Adults — Behavioral Risk Factor Surveillance System, 2011–2020. MMWR Morb Mortal Wkly Rep 2023;72:707–715. DOI: http://dx.doi.org/10.15585/mmwr.mm7226a2 .
- Bellis, MA, et al. Life Course Health Consequences and Associated Annual Costs of Adverse Childhood Experiences Across Europe and North America: A Systematic Review and Meta-Analysis. Lancet Public Health 2019.
- Adverse Childhood Experiences During the COVID-19 Pandemic and Associations with Poor Mental Health and Suicidal Behaviors Among High School Students — Adolescent Behaviors and Experiences Survey, United States, January–June 2021 | MMWR
- Hillis SD, Anda RF, Dube SR, Felitti VJ, Marchbanks PA, Marks JS. The association between adverse childhood experiences and adolescent pregnancy, long-term psychosocial consequences, and fetal death. Pediatrics. 2004 Feb;113(2):320-7.
- Miller ES, Fleming O, Ekpe EE, Grobman WA, Heard-Garris N. Association Between Adverse Childhood Experiences and Adverse Pregnancy Outcomes. Obstetrics & Gynecology . 2021;138(5):770-776. https://doi.org/10.1097/AOG.0000000000004570 .
- Sulaiman S, Premji SS, Tavangar F, et al. Total Adverse Childhood Experiences and Preterm Birth: A Systematic Review. Matern Child Health J . 2021;25(10):1581-1594. https://doi.org/10.1007/s10995-021-03176-6 .
- Ciciolla L, Shreffler KM, Tiemeyer S. Maternal Childhood Adversity as a Risk for Perinatal Complications and NICU Hospitalization. Journal of Pediatric Psychology . 2021;46(7):801-813. https://doi.org/10.1093/jpepsy/jsab027 .
- Mersky JP, Lee CP. Adverse childhood experiences and poor birth outcomes in a diverse, low-income sample. BMC pregnancy and childbirth. 2019;19(1). https://doi.org/10.1186/s12884-019-2560-8 .
- Reid JA, Baglivio MT, Piquero AR, Greenwald MA, Epps N. No youth left behind to human trafficking: Exploring profiles of risk. American journal of orthopsychiatry. 2019;89(6):704.
- Diamond-Welch B, Kosloski AE. Adverse childhood experiences and propensity to participate in the commercialized sex market. Child Abuse & Neglect. 2020 Jun 1;104:104468.
- Shonkoff, J. P., Garner, A. S., Committee on Psychosocial Aspects of Child and Family Health, Committee on Early Childhood, Adoption, and Dependent Care, & Section on Developmental and Behavioral Pediatrics (2012). The lifelong effects of early childhood adversity and toxic stress. Pediatrics, 129(1), e232–e246. https://doi.org/10.1542/peds.2011-2663
- Narayan AJ, Kalstabakken AW, Labella MH, Nerenberg LS, Monn AR, Masten AS. Intergenerational continuity of adverse childhood experiences in homeless families: unpacking exposure to maltreatment versus family dysfunction. Am J Orthopsych. 2017;87(1):3. https://doi.org/10.1037/ort0000133 .
- Schofield TJ, Donnellan MB, Merrick MT, Ports KA, Klevens J, Leeb R. Intergenerational continuity in adverse childhood experiences and rural community environments. Am J Public Health. 2018;108(9):1148-1152. https://doi.org/10.2105/AJPH.2018.304598 .
- Schofield TJ, Lee RD, Merrick MT. Safe, stable, nurturing relationships as a moderator of intergenerational continuity of child maltreatment: a meta-analysis. J Adolesc Health. 2013;53(4 Suppl):S32-38. https://doi.org/10.1016/j.jadohealth.2013.05.004 .
Adverse Childhood Experiences (ACEs)
ACEs can have a tremendous impact on lifelong health and opportunity. CDC works to understand ACEs and prevent them.
Find Info For
- Current Students
- Prospective Students
- Research and Partnerships
- Entrepreneurship and Commercialization
Quick Links
- Health and Life Sciences
- Info Security and AI
- Transformative Education
- Purdue Today
- Purdue Global
- Purdue in the News
May 24, 2024
Purdue, Google announce sustainability and AI research partnership

Purdue University and Google announced a partnership that seeks to develop innovative solutions for eco-friendly industrial buildings. Left to right: Joe Sinfield, civil engineering professor and director of Purdue’s Institute for Innovation Science; Travis Horton, civil engineering professor; Ben Townsend, Google global head of infrastructure and sustainability; Alyssa Wilcox, Purdue senior vice president of partnerships and chief of staff. (Purdue University photo provided)
Collaboration seeks to develop innovative solutions for eco-friendly industrial buildings
WEST LAFAYETTE, Ind. — Purdue University and Google announced Thursday (May 23) a new collaborative research project aimed at exploring the use of AI to develop innovative solutions for low-carbon industrial building design. The project seeks to leverage AI’s power to explore new materials, technologies and design strategies that can significantly reduce the carbon footprint of industrial buildings, such as data centers, not only across the U.S. but globally.
“Google is committed to using AI to address some of the world’s most pressing challenges, including climate change,” said Ben Townsend, global head of infrastructure and sustainability at Google. “We are excited to partner with Purdue University on this important research project, which has the potential to accelerate the adoption of low-carbon building practices in the industrial sector.”
ADDITIONAL INFORMATION
- Purdue Computes
- Purdue University to offer Google Career Certificates for learning in-demand tech skills
Conventional construction processes and physical plant operations of industrial structures pose persistent challenges when builders seek to reduce greenhouse gas emissions. That’s one reason why Purdue and Google are partnering on research efforts to develop new, sustainable building design approaches.
One focus area will be to prioritize the use of low-carbon building materials. The partners said research findings from this collaboration will be shared with the wider research community and industry stakeholders, aiming to boost the adoption of sustainable building practices.
“By combining Purdue’s expertise in engineering and building science with Google’s cutting-edge AI capabilities, we aim to develop innovative solutions that can significantly reduce the carbon footprint of industrial buildings,” said Travis Horton, a professor in Purdue’s Lyles School of Civil Engineering who also holds a courtesy appointment in mechanical engineering. “This collaboration is a testament to our commitment to sustainability and our belief in the transformative potential of AI.”
AI is a foundational component of the Institute for Physical Artificial Intelligence , a Purdue Computes initiative.
The partners said AI has the potential to have a profound impact on industrial construction, as it could provide innovative applications of low-carbon building materials, which would support reducing operational costs and making progress on global sustainability goals.
“At Google we are committed to sustainability, and we believe that technology can play a critical role in reducing carbon emissions,” said Townsend, who earned bachelor’s and master’s degrees in civil engineering from Purdue.
Google, which owns and operates data centers all over the world, is continually examining systems, including innovative construction design processes, to reduce the carbon footprint of its facilities to ensure efficiency and eco-friendly construction.
“The potential to combine Purdue’s understanding of innovation and design processes with Google’s AI capabilities to enable highly efficient and scalable sustainable design foreshadows the far-reaching promise of AI to help society address its most complex challenges,” said Joe Sinfield, a professor in Purdue’s Lyles School of Civil Engineering and the director of Purdue’s Institute for Innovation Science.
About Purdue University
Purdue University is a public research institution demonstrating excellence at scale. Ranked among top 10 public universities and with two colleges in the top four in the United States, Purdue discovers and disseminates knowledge with a quality and at a scale second to none. More than 105,000 students study at Purdue across modalities and locations, including nearly 50,000 in person on the West Lafayette campus. Committed to affordability and accessibility, Purdue’s main campus has frozen tuition 13 years in a row. See how Purdue never stops in the persistent pursuit of the next giant leap — including its first comprehensive urban campus in Indianapolis, the new Mitchell E. Daniels, Jr. School of Business, and Purdue Computes — at https://www.purdue.edu/president/strategic-initiatives .
Writer/Media contact: Wes Mills, [email protected]
Source: Travis Horton
Research News
Communication.
- OneCampus Portal
- Brightspace
- BoilerConnect
- Faculty and Staff
- Human Resources
- Colleges and Schools
Info for Staff
- Purdue Moves
- Board of Trustees
- University Senate
- Center for Healthy Living
- Information Technology
- Ethics & Compliance
- Campus Disruptions
Purdue University, 610 Purdue Mall, West Lafayette, IN 47907, (765) 494-4600
© 2015-24 Purdue University | An equal access/equal opportunity university | Copyright Complaints | Maintained by Office of Strategic Communications
Trouble with this page? Disability-related accessibility issue? Please contact News Service at [email protected] .
IMAGES
VIDEO
COMMENTS
Please use the document number 1723 to identify the guidance you are requesting. Or, contact: Office of Communication, Outreach and Development, HFM-40 Center for Biologics Evaluation and Research ...
Manufactures use the "Research Use Only" (RUO) label to declare that their products should not be used in diagnostic procedures. This enables them to avoid the time-consuming and costly documentation required for conformity-assessed in vitro diagnostic medical devices (CE-IVDs). Nevertheless, some medical laboratories, for example, still ...
Clinical laboratory professionals may not pause to remember that these labels stand for In Vitro Diagnostics (IVD) and Research Use Only (RUO). Even clinical laboratory professionals who are familiar with these regulatory designations for assays or instruments sometimes do not realize the full significance that these labels have for certain ...
Thermo Fisher Scientific GMP products can support your efforts to produce products that function consistently as intended. We follow quality standards in manufacturing, testing, documentation, and proven use. Our - CTS products, intended for use in GMP production, are manufactured at sites that are FDA registered, ISO 13485 certified, and ...
The label Research Use Only (RUO) is generally used to indicate products that are intended for scientific research only. They cannot be used for diagnostic or medical purposes. However, there is no standard definition of "research use only," and the label has slightly different meanings in the European Union and the United States.
Research Use Only (RUO) products are a distinct category of in vitro diagnostics (IVDs) exclusively tailored for laboratory research. RUOs encompass specialised reagents, equipment, and materials crucial for scientific investigations, contributing significantly to the development of cutting-edge tools and solutions for research applications.
Research product (Research Use Only/RUO) is medical device and in-vitro medical device product that is in research development stage and has not been approved to be used for clinical purposes; or which is declared RUO by the authorized body in country of origin of the manufacturer. Category 3. 1.
Commercially Distributed In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only: Frequently Asked Questions August 29, 2011 College of American Pathologists 1350 I Street, NW, Suite 590 Washington, DC 20005 (202) 354-7100 (202) 354-7155 - fax (800) 392-9994 www.cap.org
Diagnostic kits intended for diagnostic use face the full panoply of FDA regulation. In sharp contrast, research use only (RUO) products are essentially unregulated.
On June 1, 2011, the US Food and Drug Administration (FDA) 2 Office of In Vitro Diagnostic Device Evaluation and Safety issued draft guidance for industry and FDA staff intended to provide guidance regarding the FDA's thinking about in vitro diagnostic device (IVD) products labeled for "Research Use Only" (RUO) or "Investigational Use Only" (IUO) ().
The Research Use Only (RUO; 2013) and companion diagnostic (2014) guidances should be considered jointly as two parts of a coherent regulatory policy for the development of companion diagnostics. The frequently asked questions in this guidance discuss limits on use of RUO or Investigational Use Only diagnostic tests leading to clinically ...
The 21 CFR 809.10 and 21 CFR 864 define four types of IVDs: General Purpose Reagent (GPR), Investigational Use Only (IUO), Analyte Specific Reagent (ASR) and Research Use Only (RUO). This is why in the U.S., RUOs are also called RUO IVDs - In contrast, in EU, only the term RUO prevails. As per the 21 CFR, RUO products are IVD products in the ...
Label requirements are as follows: The established and proprietary names of the product, e.g., cholesterol meters; * The intended use or uses, e.g., pregnancy detection, diabetes screening, etc ...
Save for later. NEW YORK (GenomeWeb News) - The Food and Drug Administration has released a guidance document that lays out and clarifies the rules for how in vitro diagnostic products for research use only (RUO) and investigational use only (IUO) may be used, labeled, or marketed. FDA created the guidance on RUOs and IUOs, which has been ...
Research-use-only (RUO) products are intended solely for research purposes, not for diagnostic purposes. RUO products are often discussed as if they are medical devices, but since the intended use is research only, they do not fit the definition of a device and are essentially unregulated. RUO products are addressed very briefly by FDA regulations.
21 CFR Part 809.3 defines the in vitro diagnostics (IVDs) products as "reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae.Such products are intended for use in the collection, preparation, and examination of specimens taken ...
01. Introduction. This document has been developed as a result of the outcome of initial discussions on "research only products" at the Medical Devices Expert Group (MDEG) meeting of July 2003. It aims to clarify a number of issues raised by Competent Authorities with regard to products labeled as "For Research Use Only" (RUO) and their ...
Research Use Only: A non-IVD product in the laboratory research phase of development that cannot be used in diagnostic procedures. IUO: Investigational Use Only: A product being shipped or delivered for product testing and process evaluation, but not yet commercialized. EUA: Emergency Use Authorization
These recommendations can fill the gap between research use only and in vitro diagnostics. Introduction. A literature search on biomarkers and cardiovascular diseases (CVDs) highlights the poor correlation between the efforts performed in the initial steps of the development of quantitative PCR (qPCR) assay-based biomarkers, i.e., discovery and ...
The second misconception clinical laboratories should be aware of involves material labeled as Research Use Only (RUO). RUO labeling is intended for products that are still under development and are not commercially distributed. A developer would use this labeling to ship product for "investigation relating to product development" as ...
the FDA in a research context or as part of product development or a clinical trial. However, the assay cannot be used legally for clinical diagnostic procedures or any purpose other than research or investigation. The assay must be prominently labeled for Research Use Only (RUO) or Investigational Use Only (IUO) prior to shipment or delivery to a
Research Use Only Diagnostics. Staff at Catachem have been pioneers in the early development of diagnostic reagents for use on automated equipment going back to the 1970's, and the company continues to develop research versions of new reagents for future use in the human clinical laboratory. Catachem's diagnostic products have been evolving ...
The Sidney Laboratory, also known as the Centre for Plant Health, is Canada's only post-entry quarantine (PEQ), research and diagnostic facility for virus testing of all fruit-bearing trees, grapevines and small fruit (e.g. berries). PEQ facilities ensure the safe introduction of foreign plant material into Canada.
Importance Use of telehealth has increased substantially in recent years. However, little is known about whether the likelihood of completing recommended tests and specialty referrals—termed diagnostic loop closure—is associated with visit modality.. Objectives To examine the prevalence of diagnostic loop closure for tests and referrals ordered at telehealth visits vs in-person visits and ...
The Consolidated guidelines on HIV, viral hepatitis and STI prevention, diagnosis, treatment and care for key populations outline a public health response for 5 key populations (men who have sex with men, trans and gender diverse people, sex workers, people who inject drugs and people in prisons and other closed settings). They present and ...
"The dream of many research groups and companies is to implant something into the body that over the long term the body will not see, and the device can provide therapeutic or diagnostic functionality. Now we have such an 'invisibility cloak,' and this is very general: There's no need for a drug, no need for a special polymer," says ...
Leaders who are exploring how AI might fit into their business operations must not only navigate a vast and ever-changing landscape of tools, but they must also facilitate a significant cultural ...
Toxic stress, or extended or prolonged stress, from ACEs can negatively affect children's brain development, immune systems, and stress-response systems. These changes can affect children's attention, decision-making, and learning. 18. Children growing up with toxic stress may have difficulty forming healthy and stable relationships.
Purdue University and Google announced Thursday (May 23) a new collaborative research project aimed at exploring the use of AI to develop innovative solutions for low-carbon industrial building design. The project seeks to leverage AI's power to explore new materials, technologies and design strategies that can significantly reduce the carbon footprint of industrial buildings, such as data ...