- Biochemistry

Case Study #2

Related documents

Add this document to collection(s)
You can add this document to your study collection(s)
Add this document to saved
You can add this document to your saved list
Suggest us how to improve StudyLib
(For complaints, use another form )
Input it if you want to receive answer
We have a new app!
Take the Access library with you wherever you go—easy access to books, videos, images, podcasts, personalized features, and more.
Download the Access App here: iOS and Android . Learn more here!
- Remote Access
- Save figures into PowerPoint
- Download tables as PDFs

Chapter 19: Case Study: Cystic Fibrosis
Julie M. Skrzat; Carole A. Tucker
- Download Chapter PDF
Disclaimer: These citations have been automatically generated based on the information we have and it may not be 100% accurate. Please consult the latest official manual style if you have any questions regarding the format accuracy.
Download citation file:
- Search Book
Jump to a Section
Introduction.
- Examination: Age 2 Months
- Evaluation, Diagnosis, and Prognosis
- Intervention
- Conclusion of Care
- Examination: Age 8 Years
- Examination: Age 16 Years
- Recommended Readings
- Full Chapter
- Supplementary Content
C ystic fibrosis (CF) is an autosomal recessive condition affecting approximately 30,000 Americans and 70,000 people worldwide. According to the Cystic Fibrosis Foundation ( Cystic Fibrosis Foundation, 2019a ), approximately 1,000 new cases are diagnosed yearly in the United States, with a known incidence of 1 per 3,900 live births. The disease prevalence varies greatly by ethnicity, with the highest prevalence occurring in Western European descendants and within the Ashkenazi Jewish population.
The CF gene, located on chromosome 7, was first identified in 1989. The disease process is caused by a mutation to the gene that encodes for the CF transmembrane conductance regulator (CFTR) protein. This mutation alters the production, structure, and function of cyclic adenosine monophosphate (cAMP), a dependent transmembrane chloride channel carrier protein found in the exocrine mucus glands throughout the body. The mutated carrier protein is unable to transport chloride across the cell membrane, resulting in an electrolyte and charge imbalance. Diffusion of water across the cell membrane is thus impaired, resulting in the development of a viscous layer of mucus. The thick mucus obstructs the cell membranes, traps nearby bacteria, and incites a local inflammatory response. Subsequent bacterial colonization occurs at an early age and ultimately this repetitive infectious process leads to progressive inflammatory damage to the organs involved in individuals with CF.
Pop-up div Successfully Displayed
This div only appears when the trigger link is hovered over. Otherwise it is hidden from view.
Please Wait
- Publications
- Conferences & Events
- Professional Learning
- Science Standards
- Awards & Competitions
- Instructional Materials
- Free Resources
- American Rescue Plan
- For Preservice Teachers
- NCCSTS Case Collection
- Science and STEM Education Jobs
- Interactive eBooks+
- Digital Catalog
- Regional Product Representatives
- e-Newsletters
- Bestselling Books
- Latest Books
- Popular Book Series
- Prospective Authors
- Web Seminars
- Exhibits & Sponsorship
- Conference Reviewers
- National Conference • Denver 24
- Get Involved with Conferences
- Leaders Institute 2024
- National Conference • New Orleans 24
- Submit a Proposal
- Latest Resources
- Professional Learning Units & Courses
- For Districts
- Online Course Providers
- Schools & Districts
- College Professors & Students
- The Standards
- Teachers and Admin
- eCYBERMISSION
- Toshiba/NSTA ExploraVision
- Junior Science & Humanities Symposium
- Teaching Awards
- Climate Change
- Earth & Space Science
- New Science Teachers
- Early Childhood
- Middle School
- High School
- Postsecondary
- Informal Education
- Journal Articles
- Lesson Plans
- e-newsletters
- Science & Children
- Science Scope
- The Science Teacher
- Journal of College Sci. Teaching
- Connected Science Learning
- NSTA Reports
- Next-Gen Navigator
- Science Update
- Teacher Tip Tuesday
- Trans. Sci. Learning
MyNSTA Community
- My Collections
Maggie’s Illness
Protein Structure and Function in Cystic Fibrosis
By Michaela Gazdik Stofer
Share Start a Discussion

This directed case study examines the molecular basis of cystic fibrosis to emphasize the relationship between the genetic code stored in a DNA sequence and the encoded protein’s structure and function. Cystic fibrosis is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) protein that functions to help maintain salt and water balance along the surface of the lung and gastrointestinal tract. This case introduces students to “Maggie,” who has just been diagnosed with cystic fibrosis. The students must identify the mutation causing Maggie’s disease by transcribing and translating a portion of the wildtype and mutated CFTR gene. Students then compare the three-dimensional structures of the resulting proteins to better understand the effect a single amino acid mutation can have on the overall shape of a protein. Students also review the concepts of tonicity and osmosis to examine how the defective CFTR protein leads to an increase in the viscosity of mucus in cystic fibrosis patients. This case was developed for use in an introductory college-level biology course but could also be adapted for use in an upper-level cell or molecular biology course.
Download Case
Date Posted
- Generate a protein sequence through transcription and translation of a given DNA gene sequence.
- Explain the chemistry of amino acid side chains and their importance in protein folding.
- Describe how a mutation in a protein sequence leads to changes in the overall tertiary structure of the protein.
- Examine various levels of protein structure using Cn3D to view three-dimensional protein structures from NCBI’s Entrez Structure database.
- Relate the loss of function of the CFTR protein to the physiological causes of cystic fibrosis.
Protein structure; transcription; translation; DNA mutation; cystic fibrosis; genetic disease; protein function; protein folding; protein; CFTR; Cn3D
Subject Headings
EDUCATIONAL LEVEL
Undergraduate lower division, Undergraduate upper division
TOPICAL AREAS
TYPE/METHODS
Teaching Notes & Answer Key
Teaching notes.
Case teaching notes are protected and access to them is limited to paid subscribed instructors. To become a paid subscriber, purchase a subscription here .
Teaching notes are intended to help teachers select and adopt a case. They typically include a summary of the case, teaching objectives, information about the intended audience, details about how the case may be taught, and a list of references and resources.
Download Notes
Answer Keys are protected and access to them is limited to paid subscribed instructors. To become a paid subscriber, purchase a subscription here .
Download Answer Key
Materials & Media
Supplemental materials.
The following two files should be viewed with the Cn3D software to view a single domain of the CFTR and ∆F508 CFTR proteins.
You may also like
Web Seminar
Join us on Tuesday, June 4, 2024, from 7:00 PM to 8:30 PM ET, to learn about the free lesson plans and storyline units designed for high school studen...
Join us on Thursday, October 24, 2024, from 7:00 PM to 8:00 PM ET, to learn about all NSTA Teacher Awards available and how to apply.Did you come up w...

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
Margin Size
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
Case Study: Cystic Fibrosis - CER
- Last updated
- Save as PDF
- Page ID 26446
This page is a draft and is under active development.
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
Part I: A Case of Cystic Fibrosis
Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency room because she had been suffering from a chronic cough. In addition, they said that Zoey sometimes would "wheeze" a lot more than they thought was normal for a child with a cold. Upon arriving at the emergency room, the attending pediatrician noted that salt crystals were present on Zoey's skin and called Dr. Weyland, a pediatric pulmonologist. Dr. Weyland suspects that baby Zoey may be suffering from cystic fibrosis.
CF affects more than 30,000 kids and young adults in the United States. It disrupts the normal function of epithelial cells — cells that make up the sweat glands in the skin and that also line passageways inside the lungs, pancreas, and digestive and reproductive systems.
The inherited CF gene directs the body's epithelial cells to produce a defective form of a protein called CFTR (or cystic fibrosis transmembrane conductance regulator) found in cells that line the lungs, digestive tract, sweat glands, and genitourinary system.
When the CFTR protein is defective, epithelial cells can't regulate the way that chloride ions pass across cell membranes. This disrupts the balance of salt and water needed to maintain a normal thin coating of mucus inside the lungs and other passageways. The mucus becomes thick, sticky, and hard to move, and can result in infections from bacterial colonization.

- "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will die" This is an old saying from the eighteenth century and describes one of the symptoms of CF (salty skin). Why do you think babies in the modern age have a better chance of survival than babies in the 18th century?
- What symptoms lead Dr. Weyland to his initial diagnosis?
- Consider the graph of infections, which organism stays relatively constant in numbers over a lifetime. What organism is most likely affecting baby Zoey?
- What do you think is the most dangerous time period for a patient with CF? Justify your answer.
Part II: CF is a disorder of the cell membrane.
Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded. This is the challenge faced by David Gadsby, Ph.D., who for years struggled to understand the highly intricate and unusual cystic fibrosis chloride channel – a cellular doorway for salt ions that is defective in people with cystic fibrosis.
His findings, reported in a series of three recent papers in the Journal of General Physiology, detail the type and order of molecular events required to open and close the gates of the cystic fibrosis chloride channel, or as scientists call it, the cystic fibrosis transmembrane conductance regulator (CFTR).
Ultimately, the research may have medical applications, though ironically not likely for most cystic fibrosis patients. Because two-thirds of cystic fibrosis patients fail to produce the cystic fibrosis channel altogether, a cure for most is expected to result from research focused on replacing the lost channel.

5. Suggest a molecular fix for a mutated CFTR channel. How would you correct it if you had the ability to tinker with it on a molecular level?
6. Why would treatment that targets the CFTR channel not be effective for 2⁄3 of those with cystic fibrosis?
7. Sweat glands cool the body by releasing perspiration (sweat) from the lower layers of the skin onto the surface. Sodium and chloride (salt) help carry water to the skin's surface and are then reabsorbed into the body. Why does a person with cystic fibrosis have salty tasting skin?
Part III: No cell is an island
Like people, cells need to communicate and interact with their environment to survive. One way they go about this is through pores in their outer membranes, called ion channels, which provide charged ions, such as chloride or potassium, with their own personalized cellular doorways. But, ion channels are not like open doors; instead, they are more like gateways with high-security locks that are opened and closed to carefully control the passage of their respective ions.
In the case of CFTR, chloride ions travel in and out of the cell through the channel’s guarded pore as a means to control the flow of water in and out of cells. In cystic fibrosis patients, this delicate salt/water balance is disturbed, most prominently in the lungs, resulting in thick coats of mucus that eventually spur life-threatening infections. Shown below are several mutations linked to CFTR:

8. Which mutation do you think would be easiest to correct. Justify your answer. 9. Consider what you know about proteins, why does the “folding” of the protein matter?
Part IV: Open sesame
Among the numerous ion channels in cell membranes, there are two principal types: voltage-gated and ligand-gated. Voltage-gated channels are triggered to open and shut their doors by changes in the electric potential difference across the membrane. Ligand-gated channels, in contrast, require a special “key” to unlock their doors, which usually comes in the form of a small molecule.
CFTR is a ligand-gated channel, but it’s an unusual one. Its “key” is ATP, a small molecule that plays a critical role in the storage and release of energy within cells in the body. In addition to binding the ATP, the CFTR channel must snip a phosphate group – one of three “P’s” – off the ATP molecule to function. But when, where and how often this crucial event takes place has remains obscure.

10. Compare the action of the ligand-gated channel to how an enzyme works.
11. Consider the model of the membrane channel, What could go wrong to prevent the channel from opening?
12. Where is ATP generated in the cell? How might ATP production affect the symptoms of cystic fibrosis?
13. Label the image below to show how the ligand-gated channel for CFTR works. Include a summary.

Part V: Can a Drug Treat Zoey’s Condition?
Dr. Weyland confirmed that Zoey does have cystic fibrosis and called the parents in to talk about potential treatments. “Good news, there are two experimental drugs that have shown promise in CF patients. These drugs can help Zoey clear the mucus from his lungs. Unfortunately, the drugs do not work in all cases.” The doctor gave the parents literature about the drugs and asked them to consider signing Zoey up for trials.
The Experimental Drugs
Ivacaftor TM is a potentiator that increases CFTR channel opening time. We know from the cell culture studies that this increases chloride transport by as much as 50% from baseline and restores it closer to what we would expect to observe in wild type CFTR. Basically, the drug increases CFTR activity by unlocking the gate that allows for the normal flow of salt and fluids.
In early trials, 144 patients all of whom were age over the age of 12 were treated with 150 mg of Ivacaftor twice daily. The total length of treatment was 48 weeks. Graph A shows changes in FEV (forced expiratory volume) with individuals using the drug versus a placebo. Graph B shows concentrations of chloride in patient’s sweat.

14. What is FEV? Describe a way that a doctor could take a measurement of FEV.
15. Why do you think it was important to have placebos in both of these studies?
16. Which graph do you think provides the most compelling evidence for the effectiveness of Ivacafor? Defend your choice.
17. Take a look at the mutations that can occur in the cell membrane proteins from Part III. For which mutation do you think Ivacaftor will be most effective? Justify your answer.
18. Would you sign Zoey up for clinical trials based on the evidence? What concerns would a parent have before considering an experimental drug?
Part VI: Zoey’s Mutation
Dr. Weyland calls a week later to inform the parents that genetic tests show that Zoey chromosomes show that she has two copies of the F508del mutation. This mutation, while the most common type of CF mutation, is also one that is difficult to treat with just Ivacaftor. There are still some options for treatment.
In people with the most common CF mutation, F508del, a series of problems prevents the CFTR protein from taking its correct shape and reaching its proper place on the cell surface. The cell recognizes the protein as not normal and targets it for degradation before it makes it to the cell surface. In order to treat this problem, we need to do two things: first, an agent to get the protein to the surface, and then ivacaftor (VX-770) to open up the channel and increase chloride transport. VX-809 has been identified as a way to help with the trafficking of the protein to the cell surface. When added VX-809 is added to ivacaftor (now called Lumacaftor,) the protein gets to the surface and also increases in chloride transport by increasing channel opening time.

In early trials, experiments were done in-vitro, where studies were done on cell cultures to see if the drugs would affect the proteins made by the cell. General observations can be made from the cells, but drugs may not work on an individual’s phenotype. A new type of research uses ex-vivo experiments, where rectal organoids (mini-guts) were grown from rectal biopsies of the patient that would be treated with the drug. Ex-vivo experiments are personalized medicine, each person may have different correctors and potentiators evaluated using their own rectal organoids. The graph below shows how each drug works for 8 different patients (#1-#8)
19. Compare ex-vivo trials to in-vitro trials.
20. One the graph, label the group that represents Ivacaftor and Lumacaftor. What is the difference between these two drugs?
21. Complete a CER Chart. If the profile labeled #7 is Zoey, rank the possible drug treatments in order of their effectiveness for her mutation. This is your CLAIM. Provide EVIDENCE to support your claim. Provide REASONING that explains why this treatment would be more effective than other treatments and why what works for Zoey may not work for other patients. This is where you tie the graph above to everything you have learned in this case. Attach a page.
- Cystic fibrosis
Citation, DOI, disclosures and case data
At the time the case was submitted for publication Mostafa Elfeky had no financial relationships to ineligible companies to disclose.
Presentation
History of chronic sinusitis and recurrent chest infection, 2 times admission within a year. He is on prophylactic azithromycin.
Patient Data
At age of 5 years.
Increased lung volume with diffuse bronchiectatic changes , peribronchial cuffing , coarse interstitial markings, interstitial infiltrates and small patchy airspace opacities.
Bilateral dense hilum suspicious of hilar lymphadenopathy.
At age of 1 year
Diffuse ground-glass veiling on both lung fields, suggestive of pulmonary infection.
Case Discussion
Findings are in keeping with the known diagnosis of cystic fibrosis . It usually manifests with recurrent pulmonary infection .
0 public playlists include this case
Related radiopaedia articles.
- Cystic fibrosis (pulmonary manifestations)
- Pulmonary infection
Promoted articles (advertising)
How to use cases.
You can use Radiopaedia cases in a variety of ways to help you learn and teach.
- Add cases to playlists
- Share cases with the diagnosis hidden
- Use images in presentations
- Use them in multiple choice question
Creating your own cases is easy.
- Case creation learning pathway
ADVERTISEMENT: Supporters see fewer/no ads
By Section:
- Artificial Intelligence
- Classifications
- Imaging Technology
- Interventional Radiology
- Radiography
- Central Nervous System
- Gastrointestinal
- Gynaecology
- Haematology
- Head & Neck
- Hepatobiliary
- Interventional
- Musculoskeletal
- Paediatrics
- Not Applicable
Radiopaedia.org
- Feature Sponsor
- Expert advisers

- Search forums
- Cystic Fibrosis
- Thread starter wanderlost
- Start date Feb 18, 2008
- Feb 18, 2008
A friend of mine is a nursing student and has to do this case study for class, I told her i would help, but there are some things I don't know. If you know any of these answers and have the time to type them, I'd appreciate it! I already did a lot of it, but some of the transplant stuff I didn't know. But anything would help, as I will compare my answers to yours! TIA! <b>Client Profile: Erin is an 8-year-old girl who lives with her parents and two younger sisters, Rachel, who is 5 years old, and Samantha, who is 2 years old. They live in a Midwestern community where Erin's father is a bank manager and her mother is a part-time investment broker who works from home, which allows her to stay at home with the children. Both of Erin's parents are very attentive to the children and are very knowledgeable about Erin's cystic fibrosis, which was diagnosed when Erin was 3 months old. Neither of her sisters has the disease. Erin takes pancreatic enzymes with each meal and snack (six doses per day) and she performs breathing exercises twice a day. Her mother performs postural drainage 1 hour prior to breakfast, again when Erin returns from school in the afternoon, and finally each evening prior to Erin's going to bed. Case Study: During late spring Erin's breathing has become increasing congested over the past week and her parents suspect that she has developed a respiratory infection when she becomes febrile with a temperature of 37.9 0 C (100.2 0 F). They phone her pediatrician, who recommends that she be admitted to the children's hospital 20 miles away.The pediatrician calls the hospital and informs the chief respiratory resident physician of Erin's history, chief complaints at present, and impending arrival. Sputum cultures, complete blood count, serum electrolyte panel, chest x-ray, and pulmonary function diagnostics are prescribed. Erin's last admission for pulmonary clean-out was 6 months ago. Erin is admitted and her diagnostic results include hemoglobin, 18g/dL; hematocrit, 51%; white blood cell count, 15,000 cells/mm3; platelets, 250,000 cells/mm3; red blood cell count, 5.1 million cells/mm3; serum glucose, 130mg/dL; potassium, 4.0 mmol/L; sodium, 130mmol/L; chloride, 90 mmol/L; blood urea nitrogen (BUN), 26mg/dL; and creatinine, 0.7 mg/dL. Her chest x-ray shows consolidation in her right lower and middle lobes, and her oxygen saturation is 89%. Erin's pulmonary function is determined to be 45% and as you are compiling Erin's history, her mother tells you that Erin has been on the lung transplant list for 9 months. Erin weighs 44 lb on admission. Questions 1. Discuss your impressions about Erin's diagnostic values. 2. Discuss what risks Erin has for developing a pulmonary infection. 3. What pertinent information is missing? 4. Identify the common microorganisms that cause respiratory infections in a child with cystic fibrosis. 5. What is the relationship between Erin's condition and her oxygen saturation level? 6. The health care provider prescribes ceftazidime 1 g IV every 8 hours; gentamycin 50mg IV every 8 hours; and vancomycin 265 mg IV every 8 hours. Discuss why these drugs are prescribed for Erin. 7. Discuss the safety and efficacy of the doses of the antimicrobial agents prescribed for Erin. 8. What are the criteria established for lung transplant candidates? 9. Erin's condition worsens, and when no cadaver lungs are available, Erin's mother states that she wants to donate part or all of one of her lungs to Erin. After testing her mother for compatibility, the surgeon decides to proceed with the transplant the following day. Identify the priority client problems for Erin following the transplant. 10. Discuss the common immunosuppressant agents used to prevent organ rejection. 11. Erin receives a single lung transplant. Discuss what the risks are for her cystic fibrosis recurring in her transplant lung. 12. Erin and her mother recover from the surgery. Discuss the teaching priorities you will address with Erin and her parents prior to Erin's discharge. 13. What are the current statistics for successful lung transplants?</b>
Am I the only one that thinks this is a little unbelievable? 45% lung function on transplant list, at 8 years old? Anyway.. I usually frown on doing someone else's homework for them, but this seems like a fun exercise. I'm sure most of us could do this passably well, never having set foot in nursing school.. sad, eh. 1. Not too familiar with exact blood values but I *think* that hemoglobin is low and white cell count is high.. but don't quote me on that. She is well underweight for an 8 year old, and her height is not listed. 2. Risk factors? Like having CF? Or risks as in exposure to colds/illness at school? 3. Height and BMI information. GI information missing. Medication list missing. DRUG ALLERGIES missing. Any physical exam info missing - breath sounds etc, signs of dehydration, throat/nose exam, etc. 4. Staph, Pseduomonas aeuroginsa and to a lesser extent B. cepacia, Aspergillus, MRSA 5. Her 02 sucks, and she has a respiratory infection, I'm not sure what else they want you to say there. 6. Antibiotics that were shown to be useful according to her last sputum culture, I would hope. 7. That seems a bit high for her age and weight, but your friend can probably look up the specifics of that. (especially the 1g of ceftaz.. I took 2g and I weigh 3x what this imaginary patient does) 8. This is why I said it seems unreasonable. As far as I know, your FEV1 should be more in the 20-30% range.. not 45%. We have tons of people on this site in the 40's that work full time jobs and have families.. and are pre-tx. <skipping the last few since I don't have much tx knowledge> Tell your friend she needs to share the official answers with us when she gets her hw back. <img src="i/expressions/face-icon-small-tongue.gif" border="0">
- Feb 19, 2008
I too thought that the FEV1 was high for being listed on a transplant list. I am not familiar with the entire transplant procedure...I do know that chest size, blood type, antibodies, etc are significant as far as transplant goes. 1) Erin has an elevated WBC at 15,000 (normal is around 4,500 to 10,800 I think) indicates infection, her Hgb (hemaglobin) is elevated at 18 as is her Hct (hematocrit) at 51. Her RBC's are slightly elevated at 5.1. Maybe she is a bit dehydrated...but perhaps she has polycythemia as a result of chronic hypoxia (just a guess though). The consolidation in her RLL and RML indicates pneumonia. Her sodium and chloride are also low at 130 and 90 respectively also due to her CF. BUN and creat. look okay. Glucose is okay if this is not a fasting sample. She is undeweight at 44 lbs, no height listed. 3.) Alot is missing as SevenStars said: Any CT scan results , GI info (bowels, CFRD?, nutritional screens), definitely drug allergies, what organisms ultimately grew out of the sputum sample, the entire physical exam, meds, were any repeat PFT's ordered?, also..what other meds or interventions were ordered for Erin (such as O2, nutrition assessments, meds, chest physio etc...) I know I forgot other stuff... 6.) Vanco is used mainly for MRSA (and some other creepy bugs), Gent and Ceftaz are great anti pseudomonals. Hopefully this med combo was dependent on the results of her sputum culture. Gent is 3-6mg/kg/day divided equally in 3 doses usually...Erin is about 20kg, so her max dosage in a day is 120mg total. Maybe the gent is a teeny bit too high. The vanco dosage looks okay. I have seen 1g Ceftazadime given q8 in young patients. I am at loss with the xplant questions.. 11.) The transplanted lung won't develop CF in them...rejection maybe, but not CF..the DNA of the new lung does not have CF Wish I could be more helpful...Hugs, Jenn <img src="i/expressions/face-icon-small-smile.gif" border="0">
[No message]
- This site uses cookies to help personalise content, tailor your experience and to keep you logged in if you register. By continuing to use this site, you are consenting to our use of cookies. Accept Learn more…

How I learned to become a patient care advocate for my daughter
A difficult period in this columnist's life taught her a valuable lesson

by Jennifer Chamberlain | May 28, 2024
Share this article:

For days after having an anatomy scan , I spent most of the time lying on the couch, crying. My eyes were so swollen I had to ice them to keep them open.
We were living every parent’s worst nightmare: Our unborn baby had ultrasound abnormalities. The fear of the unknown consumed me. While I’d struggled with anxiety, dealing with a high-risk pregnancy took it to another level.
Days after receiving the news, my friend persuaded me to attend a Fourth of July party. I badly wanted to feel normal and act like none of this was happening, but I couldn’t escape my ruminating thoughts. While everyone celebrated the holiday, I hid in a room having a panic attack.
I knew I needed help. I started seeing a reproductive psychiatrist , who consulted with my obstetrician and perinatologist . She prescribed me an antidepressant, explaining that the stress I was under could be more harmful than the potential side effects of the medication.

How my daughter and I use mindfulness for anxiety, and life
While my husband and I had been confirmed only as cystic fibrosis (CF) carriers , my instincts were strong; I knew my unborn baby had the disease. And one of the potential side effects of the medication could affect our unborn baby’s lungs. The thought of taking an antidepressant terrified me. I had to decide whether to take it to help myself while potentially putting my baby at risk for lung complications.
I needed time and help to process my emotions. I had several sessions with my reproductive psychiatrist and a maternal health counselor to help me make a decision. But I didn’t feel truly heard, which heightened my anxiety.
A lie and a lesson
So I lied to everyone — my doctors, my parents, and even my husband. I went as far as picking up the prescription and setting it on my nightstand so that everyone would think I was taking it.
Around that time, I began to come out of the shock on my own. I started learning more about cystic fibrosis and medical advancements. My anxiety and depression waned. Everyone thought the medication was working. I felt like the biggest sham.
Of course, I don’t recommend that anyone lie, especially to their medical team. But I think my story affirms that a patient’s voice needs to be heard, whether in relation to a physical or mental health issue. I wasn’t vocal enough about my concerns and felt ashamed for questioning the opinions of medical professionals. What I failed to recognize was that patients are most affected by treatment decisions. Doctors make recommendations, but patients must live with the decisions.
Today, I’m patient advocate for my 5-year-old daughter, Claire, and will be until she can articulate her needs on her own. In that role, I’m often put in a position of deferring to medical professionals. My own experience provided me a new perspective on how to vocalize concerns and advocate for exploring all treatment options, whether for myself or for Claire.
I now ask questions and try not to leave the clinic without answers. This choice can be uncomfortable at times, but I know how important it is for Claire to understand that her voice should always heard.
Note: Cystic Fibrosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis , or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. The opinions expressed in this column are not those of Cystic Fibrosis News Today or its parent company, BioNews, and are intended to spark discussion about issues pertaining to cystic fibrosis.
About the Author

Leave a comment
Fill in the required fields to post. Your email address will not be published.
Your CF Community

Recent Posts
- How I learned to become a patient care advocate for my daughter May 28, 2024
- Rat fetuses don’t appear harmed by giving pregnant mothers Trikafta May 28, 2024
- I’m finally giving myself permission to live fully again May 27, 2024
- A letter to my younger self offers wisdom gleaned from experience May 24, 2024
- Kaftrio leads to small gains in exercise capacity after year: Study May 24, 2024
Recommended reading

Rat fetuses don’t appear harmed by giving pregnant mothers Trikafta

I’m finally giving myself permission to live fully again

A letter to my younger self offers wisdom gleaned from experience
Subscribe to our newsletter.
Get regular updates to your inbox.
Case Study - What is the Relationship Between the Cell Membrane and Cystic Fibrosis?
CF affects more than 30,000 kids and young adults in the United States. It disrupts the normal function of epithelial cells — cells that make up the sweat glands in the skin and that also line passageways inside the lungs, pancreas, and digestive and reproductive systems.
The inherited CF gene directs the body's epithelial cells to produce a defective form of a protein called CFTR (or cystic fibrosis transmembrane conductance regulator) found in cells that line the lungs, digestive tract, sweat glands, and genitourinary system.
When the CFTR protein is defective, epithelial cells can't regulate the way that chloride ions pass across cell membranes. This disrupts the balance of salt and water needed to maintain a normal thin coating of mucus inside the lungs and other passageways. The mucus becomes thick, sticky, and hard to move, and can result in infections from bacterial colonization.

1. "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will die"
This is an old saying from the eighteenth century and describes one of the symptoms of CF (salty skin). Why do you think babies in the modern age have a better chance of survival than babies in the 18th century?
2. What symptoms lead Dr. Weyland to his initial diagnosis?
3. Consider the graph of infections, which organism stays relatively constant in numbers over a lifetime?
What organism is most likely affecting baby Zoey?
4. Explain how the CF gene affects the cell membrane.
5. Consider what you know about TONICITY and the cell membrane. Why is it important to regulate salt in cells?
Part II: CF is a disorder of the cell membrane.
Imagine a door with key and combination locks on both sides, back and front. Now imagine trying to unlock that door blind-folded. This is the challenge faced by David Gadsby, Ph.D., who for years struggled to understand the highly intricate and unusual cystic fibrosis chloride channel – a cellular doorway for salt ions that is defective in people with cystic fibrosis.
His findings, reported in a series of three recent papers in the Journal of General Physiology, detail the type and order of molecular events required to open and close the gates of the cystic fibrosis chloride channel, or as scientists call it, the cystic fibrosis transmembrane conductance regulator (CFTR).
Ultimately, the research may have medical applications, though ironically not likely for most cystic fibrosis patients. Because two-thirds of cystic fibrosis patients fail to produce the cystic fibrosis channel altogether, a cure for most is expected to result from research focused on replacing the lost channel.

6. Compare the normal and the mutant CFTR protein. How would you correct the mutant protein if you had the ability to tinker with it on a molecular level?
7. Why would treatment that targets the CFTR channel not be effective for ⅔ of those with cystic fibrosis? 8. Sweat glands cool the body by releasing perspiration (sweat) from the lower layers of the skin onto the surface. Sodium and chloride (salt) help carry water to the skin's surface and are then reabsorbed into the body. Why does a person with cystic fibrosis have salty tasting skin?
Part III: No cell is an island
Like people, cells need to communicate and interact with their environment to survive. One way they go about this is through pores in their outer membranes, called ion channels, which provide charged ions, such as chloride or potassium, with their own personalized cellular doorways. But, ion channels are not like open doors; instead, they are more like gateways with high-security locks that are opened and closed to carefully control the passage of their respective ions.

9. Which mutation do you think would be easiest to correct? Justify your answer.
10. Consider what you know about proteins, why does the "folding" of the protein matter?
Part IV: Open Sesame

Among the numerous ion channels in cell membranes, there are two principal types: voltage-gated and ligand-gated. Voltage-gated channels are triggered to open and shut their doors by changes in the electric potential difference across the membrane. Ligand-gated channels, in contrast, require a special “key” to unlock their doors, which usually comes in the form of a small molecule.
CFTR is a ligand-gated channel, but it’s an unusual one. Its “key” is ATP, a small molecule that plays a critical role in the storage and release of energy within cells in the body. In addition to binding the ATP, the CFTR channel must snip a phosphate group – one of three “P’s” – off the ATP molecule to function. But when, where and how often this crucial event takes place has remained obscure.
11. Label the image to the right to show how the ligand-gated channel for CFTR works. (Structures: Ligand-gated channel protein, ATP, phospholipids). Summarize how this channels works.
12. Where is ATP generated in the cell? How might ATP production affect the symptoms of cystic fibrosis?
Part V: Can a Drug Treat Zoey's Condition?
Dr. Weyland confirmed that Zoey does have cystic fibrosis and called the parents in to talk about potential treatments. “Good news, there are two experimental drugs that have shown promise in CF patients. These drugs can help Zoey clear the mucus from her lungs. Unfortunately, the drugs do not work in all cases.” The doctor gave the parents literature about the drugs and asked them to consider signing Zoey up for trials.
The Experimental Drugs
Ivacaftor ™ is a potentiator that increases CFTR channel opening time. We know from the cell culture studies that this increases chloride transport by as much as 50% from baseline and restores it closer to what we would expect to observe in wild type CFTR. Basically, the drug increases CFTR activity by unlocking the gate that allows for the normal flow of salt and fluids.
In early trials, 144 patients all of whom were over the age of 12 were treated with 150 mg of Ivacaftor twice daily. The total length of treatment was 48 weeks. Graph A shows changes in FEV (forced expiratory volume) with individuals using the drug versus a placebo. Graph B shows concentrations of chloride in patient’s sweat.

13. What is FEV (if you're not sure, look this one up)? Describe a way that a doctor could take a measurement of FEV.
14. Why do you think it was important to have placebos in both of these studies?
15. Which graph do you think provides the most compelling evidence for the effectiveness of Ivacaftor. Defend your choice.
16. Take a look at the mutations that can occur in the cell membrane protein from Part III. For which mutation do you think Ivacaftor will be most effective. Justify your answer.
17. Would you sign Zoey up for clinical trials based on the evidence? What concerns would a parent have before considering an experimental drug?
Part VI: Zoey's Mutation
Dr. Weyland calls a week later to inform the parents that genetic tests show that Zoey chromosomes show that she has two copies of the F508del mutation. This mutation, while the most common type of CF mutation, is also one that is difficult to treat with just Ivacaftor. There are still some options for treatment.
In people with the most common CF mutation, F508del, a series of problems prevents the CFTR protein from taking its correct shape and reaching its proper place on the cell surface. The cell recognizes the protein as not normal and targets it for degradation before it makes it to the cell surface. In order to treat this problem, we need to do two things: first, an agent to get the protein to the surface, and then ivacaftor (VX-770) to open up the channel and increase chloride transport. VX-809 has been identified as a way to help with the trafficking of the protein to the cell surface. When added VX-809 is added to ivacaftor (now called Lumacaftor,) the protein gets to the surface and also increases in chloride transport by increasing channel opening time.
In early trials, experiments were done in-vitro, where studies were done on cell cultures to see if the drugs would affect the proteins made by the cell. General observations can be made from the cells, but drugs may not work on an individual’s phenotype. A new type of research uses ex-vivo experiments, where rectal organoids (mini-guts) were grown from rectal biopsies of the patient that would be treated with the drug. Ex-vivo experiments are personalized medicine, each person may have different correctors and potentiators evaluated using their own rectal organoids.
The graph below shows how each drug works for 8 different patients (#1-#8). Swelling in the organoid indicates the the channels within the cell membrane are allowing material to pass.

19. . Compare ex-vivo trials to in-vitro trials.
20. One the graph, label the group that represents Ivacaftor and Lumacaftor. What is the difference between these two drugs?
21. Complete a CER Chart.
If the profile labeled #7 is Zoey, rank the possible drug treatments in order of their effectiveness for her mutation. This is your CLAIM. Provide EVIDENCE to support your claim Provide REASONING that explains why this treatment would be more effective than other treatments and why what works for Zoey may not work for other patients. This is where you tie the graph above to everything you have learned in this case. Attach a page.

Source & Credits
- "CFTR Protein Panels" by Lbudd14 - Own work. Licensed under Creative Commons Attribution-Share Alike 3.0 via Wikimedia Commons
- http://newswire.rockefeller.edu/2003/12/19/scientists-finally-pry-stubborn-cellular-door-ajar/
- http://en.wikipedia.org/wiki/Cystic_fibrosis
- http://www.medscape.org/viewarticle/806649_transcript
- http://www.cff.org/research/clinicalresearch/faqs/combinedkalydeco-vx-809/#Expanded-Access
- Ifacaftor Trial Graph: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3230303/
- Organoid swelling graph: http://www.potentiate.info/?q=trio-clinical-trial-ivacaftor-genistein
- Life expectancy graph: http://www.nationaljewish.org/healthinfo/conditions/cysticfibrosis/life-expectancy/
Follow-up Article: What it's like to have two kids with cystic fibrosis More information at John Hopkins Cystic Fibrosis Center
Other Resources on Cystic Fibrosis
Cystic Fibrosis Mutations Cell Membrane and Transport (Slides)
Pursuit home
- All sections
A new study hopes to find out whether cystic fibrosis could be treated in utero
Medications for cystic fibrosis are currently only approved for children over two, but new research aims to guide the safe use of treatment during pregnancy and breastfeeding
By Dr Elena Schneider-Futschik, University of Melbourne
Just five years ago, starting a family was almost unheard of for patients with cystic fibrosis (CF). And for expectant parents, it was often a huge shock when their child received a diagnosis of CF.
Affecting around 100,000 people worldwide , including 3,700 Australians, CF is an inherited disorder where one in 25 people carry the altered gene. When a person has two copies of that gene, the body’s mucus and sweat become very thick and salty, severely damaging the lungs and digestive system.
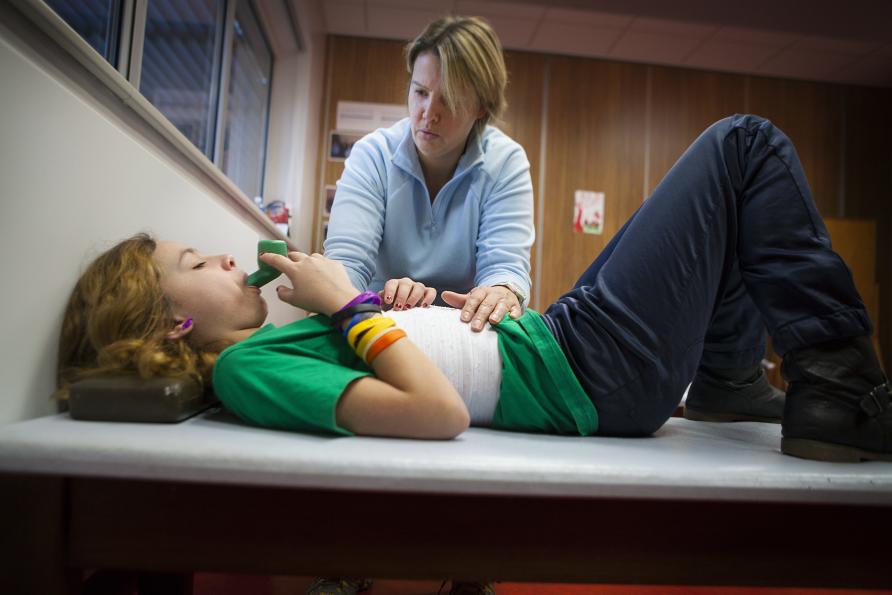
As there is currently no cure for CF, people with the condition require intensive daily physiotherapy to clear the lungs and airways, countless medications and frequent hospitalisations.
Thankfully, the landscape of CF is now dramatically changing.
In the last 10 years, the predicted life expectancy of patients with CF has increased from 40 years of age to 71.6 years – if Trikafta drug therapy is started during adolescence.
The success of Trikafta and other medications also means more people with CF are becoming parents for the first time.

Bringing answers to children with a little-known genetic condition
But as well as bringing joy, this creates a challenge. Because CF treatments are currently only approved for children over two years old, people are seeking much-needed guidance on how to proceed safely with these drugs during pregnancy and breastfeeding.
Our latest animal study found that CF drugs cross the placenta and accumulate in fetal tissues.
We also showed for the first time that entry of Trikafta to the developing brain is not restricted by fetal blood-brain barriers (BBB) – a network of blood vessels and tissue that allows water and oxygen to pass into the brain but keeps out bacteria and some drugs.
To understand more about how Trikafta impacts human fetal development, we are now embarking on a study that includes women and babies – with and without CF – to better guide treatment.

PREGNANCY AND CF
Trifkafta is part of the highly effective modulator therapies (HEMTs), which correct the underlying genetic change that cause CF disease, reducing effects on the lungs and digestive system.
In the clinical trials of Trikafta, pregnant women were excluded as it was expected that women with CF would not fall pregnant because of the effects of the condition and their shorter life expectancy.
However, this is not something we see in the clinic.
The number of pregnancies in the US has doubled from around 300 in 2019 to almost 700 after the approval of Trikafta.
Living with a rare genetic disorder
In Australia, Trikafta was added to the Pharmaceutical Benefits Scheme (PBS) in March 2022, and since then the number of pregnancies in mothers with CF has risen steadily to around 60 in 2023 .
During this time, a handful of clinical reports from doctors also identified that all components of HEMT pass through the placenta .
So patients currently face a huge dilemma, if they stop drug treatment, their own lung function decreases again, putting them and their baby at higher risk for preterm delivery.
But Trikafta is currently only approved by the US Food and Drug Administration Food and Drug Administration (FDA) for use in children aged two and over.
So on the one hand, if a mother with CF takes Trikafta, the unborn child shouldn’t be exposed to unnecessary drugs, as the effects are still unknown. On the other hand, an unborn child with CF might benefit significantly from early exposure, potentially preventing some of the irreversible symptoms like male infertility that begin before birth.

With 90 per cent of CF patients estimated to be on HEMTs by 2025, families and clinicians urgently need solid information from larger studies to make informed decisions.
We need to avoid families feeling that their only option is to use CF medication outside current recommendations .
DEVELOPING BETTER GUIDANCE FOR PARENTS
Building on the individual clinical case reports, our latest research showed that when Trikafta is taken during pregnancy, it does cross the placenta in animal models, accumulating in key cystic fibrosis tissues including the pancreas, liver, lungs and the brain.

Childcare centres and car exhaust: A dangerous mix
This is a significant finding, firstly because it highlights the need for long-term follow-up for babies with early exposure to Trikafta, but it also offers a promising new avenue of treatment. If safe, the accumulation of Trikafta in the baby’s body could help to reduce the early effects of CF.
But, we are still missing larger data sets to provide the best guidance for patients.
With this in mind, our team has two major studies underway.
The first is in collaboration with Associate Professor Martin Donnelley ’s lab at the University of Adelaide to look at the benefits and potential risks of early life exposure to Trikafta.
This project will measure levels of Trikafta during pregnancy for both the mother and baby. Studying this in mothers with and without CF and their babies will give us a much-needed understanding of how Trikafta is absorbed and moves across the placenta.
We want to understand whether drug exposure early in life is safe for women with CF and their children, and whether it leads to prevention or delay in the onset of CF symptoms.

For the second study, we’ve partnered with Professor Jen Taylor-Cousar from the University of Colorado. Professor Taylor-Cousar leads the Maternal and Fetal Outcomes in the Era of Modulators (Mayflowers) trial – the first to evaluate changes in lung function in women with CF who are taking HEMTs during pregnancy and for two years after.
Closer to a cure
Our aim with this research is to add substantial knowledge on maternal and infant CF health , providing broad and long-term safety data for the use of Trikafta in pregnancy.
Asthma and staying out of hospital
But importantly, the model adds another, even more, promising dimension: If Trikafta is safe during pregnancy, does early exposure to these drugs delay the onset of symptoms for the baby?
This could equate to something close to a cure.
Positive outcomes would represent a paradigm shift in the way HEMTs are prescribed during pregnancy and how cystic fibrosis – and potentially other diseases – are treated in utero.
Ultimately, we want to translate our findings into treatments that arrest the development and progression of CF symptoms, translating this knowledge into clinical guidelines.
Our end goal would be to change the labelling of Trikafta for pregnant women and children under two years of age.
Importantly, this research will also offer women living with cystic fibrosis a sense of normality - a normal pregnancy and all the wonderful experiences that it brings.
Please see your healthcare provider for individual advice on CF medications.
Elena Schneider-Futschik’s research is a partnership with Professor Jen Taylor-Cousar (lead of the Mayflowers trial) and the Australian CF Data Registry. This work is supported by a US Cystic Fibrosis Foundation grant, Cure4CF Impact Grant, the Bellberry New Investigator Award, and the Japanese Respiratory Society ECR Development Award. Cystic Fibrosis Australia awarded PhD student Danni Li the CFWA Post Graduate Top Up Scholarship 2024.
Banner: iStock
- Open access
- Published: 28 May 2024
The long-term effect of elexacaftor/tezacaftor/ivacaftor on cardiorespiratory fitness in adolescent patients with cystic fibrosis: a pilot observational study
- Nela Stastna 1 , 2 ,
- Lenka Hrabovska 3 ,
- Pavel Homolka 3 ,
- Lukas Homola 4 ,
- Michal Svoboda 5 ,
- Kristian Brat 6 &
- Libor Fila 1
BMC Pulmonary Medicine volume 24 , Article number: 260 ( 2024 ) Cite this article
Metrics details
Physical activity is a crucial demand on cystic fibrosis treatment management. The highest value of oxygen uptake (VO 2peak ) is an appropriate tool to evaluate the physical activity in these patients. However, there are several other valuable CPET parameters describing exercise tolerance (W peak , VO 2VT1 , VO 2VT2, VO 2 /HR peak , etc.), and helping to better understand the effect of specific treatment (V E , V T , V D /V T etc.). Limited data showed ambiguous results of this improvement after CFTR modulator treatment. Elexacaftor/tezacaftor/ivacaftor medication improves pulmonary function and quality of life, whereas its effect on CPET has yet to be sufficiently demonstrated.
We performed a single group prospective observational study of 10 adolescent patients with cystic fibrosis who completed two CPET measurements between January 2019 and February 2023. During this period, elexacaftor/tezacaftor/ivacaftor treatment was initiated in all of them. The first CPET at the baseline was followed by controlled CPET at least one year after medication commencement. We focused on interpreting the data on their influence by the novel therapy. We hypothesized improvements in cardiorespiratory fitness following treatment. We applied the Wilcoxon signed-rank test. The data were adjusted for age at the time of CPET to eliminate bias of aging in adolescent patients.
We observed significant improvement in peak workload, VO 2 peak , VO 2VT1 , VO 2VT2 , V E /VCO 2 slope, V E , V T , RQ, VO 2 /HR peak and RR peak. The mean change in VO 2 peak was 5.7 mL/kg/min, or 15.9% of the reference value (SD ± 16.6; p = 0.014). VO 2VT1 improved by 15% of the reference value (SD ± 0.1; p = 0.014), VO 2VT2 improved by 0.5 (SD ± 0.4; p = 0.01). There were no differences in other parameters.
Exercise tolerance improved after elexacaftor/tezacaftor/ivacaftor treatment initiation. We suggest that the CFTR modulator alone is not enough for recovering physical decondition, but should be supplemented with physical activity and respiratory physiotherapy. Further studies are needed to examine the effect of CFTR modulators and physical therapy on cardiopulmonary exercise tolerance.
Peer Review reports
Cystic fibrosis (CF) is the genetic disorder affecting the lungs worldwide. There is substantial heterogeneity of clinical manifestation in patients with CF. Cystic fibrosis transmembrane conductance regulator (CFTR) mutation results in the development of bronchiectasis, recurrent infectious exacerbations and lung function decline. Thus, patients moreover have increased dead space (V D ), which is why the alveolar ventilation is lower compared to healthy subjects. During exercise in CF patients, tidal volume (V T ) increases inadequately, the respiratory rate (RR) then increases to heighten minute ventilation (V E ) and reach an adequate oxygen uptake (VO 2 ). Response to physical activity is inefficient [ 1 , 2 ]. Muscle function and muscle mass, the same as cardiac abnormalities, are mostly limitations in mild or moderate CF, patients with severe lung disease, oxygen delivery and non-physiological respiratory mechanics limit exercise capacity [ 1 , 3 , 4 , 5 , 6 , 7 ].
Cardiopulmonary exercise testing (CPET) measures aerobic exercise capacity and provides a comprehensive assessment of respiratory, cardiac and musculoskeletal function during exercise and recovery and may be used for prognosis and risk assessment [ 8 ]. Not just VO 2 peak is a predictor of survival, but also peak workload (W peak), V E /VO 2 peak (ventilatory equivalent for oxygen, EQO 2 ) and V E /VCO 2 peak (ventilatory equivalent for carbon dioxide, EQCO 2 ) may be significant [ 8 ].
Ivacaftor and lumacaftor/ivacaftor combination had no effect on VO 2 peak, only sets of case reports in tezacaftor/ivacaftor demonstrated minor improvements [ 9 , 10 , 11 , 12 , 13 ]. The novel triple combination of the modulators of CFTR channel, elexacaftor/tezacaftor/ivacaftor (ETI) provides substantial clinical improvement and prolonged median predicted survival, improves lung functions (ppFEV 1 increases by 14.3%); also promotes over-nutrition and overweight, which might affect the physical activity attitude otherwise [ 14 , 15 ]. The exact quantitative evaluation is still not well known because there is not enough documented evidence of CPET-derived measures of triple combination. The data published so far are not sufficiently convincing. ETI seem to improve VO 2 peak, but still lack more detailed data about the physical tolerability mechanism [ 16 ]. By performing this trial, we try to fill the gap in the knowledge of additional CPET parameters, than just VO 2 peak. These are important tools to evaluate the effect of the new treatment on prognosis and survival.
We hypothesize that patients with CF will improve their physical activity by several adaption mechanisms, e.g. higher myocardial contractility and cardiac output (HR, higher AT which correlate with decreasing values of RQ and V E /VO 2 ), more effective gas exchange (increased VO 2 peak and VCO 2 ), lower work of breathing (improved V E due to increased V T , RR and decreased V D /V T ), and lower hyperventilation following elexacaftor/tezacaftor/ivacaftor use. Also, the aim of this study is to define other CPET parameters suitable for evaluating the cardiorespiratory demands in cystic fibrosis.
This is a prospective observational, non-randomised study. Inclusion criteria comprised a diagnosis of CF based on current guidelines, EMA-approved genotype for ETI indication and informed consent provided by the patient or their legal representative. All the patients were ETI treatment-naïve. For this study, we analysed data of patients with CF aged 8–19 years at the time of the first testing, who had a full CPET meeting between January 23, 2019, and February 9, 2023. The follow-up measurement was performed at least 12 months after ETI commencement (Kaftrio ®, elexacaftor 100 mg, tezacaftor 50 mg, ivacaftor 75 mg; always used in combination with Kalydeko ®, ivacaftor 150 mg). This was the only intervention made; patients haven’t engaged in standardized exercise training.
All the testing was performed during a clinically stable period. We used an exhaustive ramp incremental (10-25W/min) cycling CPET (Ergoline, Ergoselect, Bitz, Germany) protocol. We selected the ramp protocol based on the patient’s physical activity level, body weight and sex. After a 3-min warm-up (10-40W), all the participants completed a test to the point of exhaustion. The protocol was tailored to the individual to yield a fatigue-limited exercise duration of 8–12 min. A five-minute active cool down period followed CPET. Breath-by-breath analysis was provided, and the O 2 and CO 2 concentrations of exhaled air with ventilatory volume was measured via face mask with connected gas and flow spirometer sensors. The stress test was performed on the ergometer ERGOLINE, and the exhaled gases was analysed by POWER CUBE – Schiller (Switzerland). A ramp protocol was used while VO 2 , VO 2VT1 , VO 2VT2 , V E /VCO 2 , V E /VO 2 , V E /VCO 2 slope, V E , V T , RQ, VO 2 /HR, RR parameters were measured every 10 s, and peak values taken as the highest 15 s achieved during the test. Blood pressure and Sp0 2 were measured during CPET monitoring.
We evaluated anthropometric parameters: height (cm), weight (kg). Our outcome was to evaluate respiratory CPET-derived parameters: maximal workload (W, W/kg, % ref.), RQ max., VO 2 peak (L, mL/kg/min, % ref.,), VO 2VT1 (L/min, % ref.), VO 2VT2 , (L/min), V E /VCO 2 slope, V E (L/min, % ref.), V T (L, % ref.), V D /V T in rest and in maximal effort, RR (min −1 ), VO 2 /HR (ml/beats per minute).
Statistical methods
Numerical parameters are described by mean (standard deviation = SD). Change during two times is tested by paired Wilcoxon signed-rank test. All tests are two-sided on level of significance 5%. Analysis was prepared in the software R (v4.2) (Bell Laboratories, Inc., Windsor, WI, US).
Thirteen patients were eligible, but only 10 patients with CF met the criteria for ETI therapy and were included in the final analysis. The mean age was 14 years, mean ppFEV 1 89.4%, 70% of the patients with CF were F508del homozygotes. Baseline patients’ characteristics are presented in Table 1 .
We observed significant improvements in VO 2 peak , the mean change was 0.8 L (SD ± 0.6; p = 0.002), 15.9% of the reference value (SD ± 16.6; p = 0.014). As well as VO 2VT1 improved by 15% of the reference value (SD ± 0.1; p = 0.014) and VO 2VT2 improved by 0.5 (SD ± 0.4; p = 0.01). Patients achieved also better VO 2VT2 , VE/VCO 2 slope, V E , V T , RQ, VO 2 /HR peak , and RR peak values. Even V D /V T marker improved. Only RQ remained unchanged.
Complete data are presented in Table 2 . The parameters were recalculated based on age and current weight (% ref.), so the impact of aging was eliminated.
During controlled CPET, all the patients indicated the fatigue of lower extremities as their reason for stopping. We did not observe any exercise-induced arrythmia.
In this study, we report improvement in most of the parameters, which are valuable predictors of death or lung transplant in CF (VO 2 peak, max. effort, peak work rate, V E /VCO 2 slope) and parameters valuable to understand the ventilatory efficiency (VO 2VT1 , VO 2VT2 , V E , V T , V D /V T VO 2 /HR peak and RR peak) [ 8 ]. An abnormally low exercise capacity and deconditioning in CF results in VO 2 peak < 82% predicted and/or peak workload < 93% predicted, VO 2VT1 occurring < 50% predicted VO 2 peak [ 17 ]. Patients in this cohort achieved improvement in two of these parameters, beyond deconditioning. This might suggest improved prognosis in patients with CF treated with ETI for at least one year.
These data provided on triple combination of CFTR modulators therapy are higher than those achieved on double combination (lumacaftor/ivacaftor or tezacaftor/ivacaftor) in Danish patients with CF followed for the same period of use (VO 2 peak 1.07 mL/min/kg, maximal workload change 14.2W) [ 18 ]. Therefore, ETI might be more effective in improving CPET-derived parameters, but improvement is likely multifactorial, and further investigation in a larger patient cohort is necessary.
Older patients with CF aged > 40 years deal with specific comorbidities, while younger patients with CF are healthier than ever due to the variant treatment strategies. Still, physical fitness in CF takes an indisputable position to effect quality of life and prognosis. Medication which reduces the amount of mucus in the respiratory tract and improve pulmonary function could not be the only reason for improved exercise tolerance. Patients eligible into our study were mostly teenagers; the maximal VO 2 peak increased significantly in absolute but also in relative values, leaving minimal doubts about the role of ageing bias over the study period [ 17 ]. Other CPET variables, VO 2VT1 and VO 2VT2 , demonstrate improved aerobic capacity, physical fitness, and more effective training to the maximal effort. Improvement of V D /V T suggests more effective ventilation and decreased V/Q mismatch, as well as improved lung function in general. This would also hint at statistically significant improvement in VO 2 /HR.
In this cohort, the mean ppFEV 1 is 89.4%; research suggests that in mild lung disease, the respiratory limitation of exercise capacity is rather low [ 13 ]. Pulmonary function improvement alone in CFTR modulator users would then be unlikely to change the exercise tolerance. The impact of ETI on the exercise tolerance improvement is hypothesized by several mechanisms [ 19 ]. CFTR protein is expressed in myocardial cells, vascular smooth muscle cells, and sarcolemma and sarcoplasm of skeletal muscle cells [ 20 , 21 , 22 ]. Impaired CFTR function results in local vasoconstriction and affects nitric oxide production [ 23 ].Therefore, the CFTR modulators might improve reduced peripheral O 2 extraction during exercise and utilisation of O 2 not only by skeletal muscle, but it is also suggested, by reduced V E / VO 2 peak [ 16 ]. CFTR protein is hypothesized to be involved in regulating mitochondrial oxidative stress and mitochondrial function in adenosine triphosphate production [ 24 , 25 ].
Decreased systemic inflammation by reducing several interleukins and pro-inflammatory mediators after CFTR modulator use affects cardiorespiratory fitness. Inter alia, chronic inflammation (especially Pseudomonas aeruginosa airway infection) relates to impaired aerobic capacity [ 26 , 27 ]. Systemic inflammation is demonstrated to lead to muscle atrophy and impaired contractility [ 28 ].
To examine body composition change, it is necessary to distinguish the mechanism of VO 2 peak change. The pattern of weight gain (whether muscle or fat) due to triple therapy must be studied. This is because increased adiposity has been suggested to contribute to the decreased VO 2 outcome [ 19 ]. Even in this cohort, the patient who gained the most weight (+ 20.4 kg), where BMI increased from 21.86 kg/m 2 to 28.38 kg/m 2 , reported the highest VO 2 peak decrease (41.9 mL/kg/min to 35.2 mL/kg/min). Last but not least, it is necessary to consider the change in the mental state of patients on triple therapy and the awareness of new life horizons and possibilities, along with the awareness of the need for more intensive care for overall fitness.
There are several limitations of this study. First, the baseline testing was performed during the COVID-19 pandemic era, so we were not able to recruit more patients in this trial. Even a change in the patient’s physical activity manners during the pandemic era changed to a more sedentary style, which is difficult to quantify. Second, we did not perform a capillary blood gas analysis, and we were therefore not able to assess the V/Q mismatch. Third, the increase in absolute values of certain parameters (e.g. VO 2 peak) might partly be attributed to the given adolescent’s growth. However, there were significant improvements also in relative values (% of predicted), therefore aging bias (if any) appears to be limited. Comparative trials with a cohort of same-aged CF patients ineligible for ETI would reveal other perspectives, and the risk of a worsened overall status due to severe CFTR pathogenic variants could bias the results.
Conclusions
We demonstrated improvements in cardiorespiratory fitness in adolescent patients with cystic fibrosis following at least one year of ETI therapy by performing controlled CPET testing. CFTR modulator treatment alone might not be effective in transforming all the mechanisms of exercise intolerance. Understanding the impact of new therapeutical strategies in cystic fibrosis is important for better therapeutical evaluation and survival assessment. Further comparative trials with a larger cohort need to be performed to streamline our results.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
Anaerobic threshold
Body mass index
Cystic fibrosis transmembrane conductance regulator
- Cystic fibrosis
Carbon dioxide
Coronavirus disease of 2019
- Cardiopulmonary exercise testing
Ventilatory equivalent for oxygen
Ventilatory equivalent for carbon dioxide
- Elexacaftor/tezacaftor/ivacaftor
Probability value
Percent predicted forced expiratory volume in one second
Revolution per minute
Respiratory quotient
Respiratory rate
Standard deviation
Ventilation/perfusion
Carbon dioxide elimination
Minute ventilation
Tidal volume
Oxygen uptake
Oxygen uptake on the first ventilatory threshold
Oxygen uptake on the second ventilatory threshold
Cerny FJ, Pullano TP, Cropp GJ. Cardiorespiratory adaptations to exercise in cystic fibrosis. Am Rev Respir Dis. 1982;126(2):217–20.
CAS PubMed Google Scholar
Thin AG, Dodd JD, Gallagher CG, Fitzgerald MX, Mcloughlin P. Effect of respiratory rate on airway deadspace ventilation during exercise in cystic fibrosis. Respir Med. 2004;98(11):1063–70.
Article CAS PubMed Google Scholar
Regnis JA, Donnelly PM, Robinson M, Alison JA, Bye PT. Ventilatory mechanics at rest and during exercise in patients with cystic fibrosis. Am J Respir Crit Care Med. 1996;154(5):1418–25.
Nixon PA, Joswiak ML, Fricker FJ. A six-minute walk test for assessing exercise tolerance in severely ill children. J Pediatr. 1996;129(3):362–6.
Pastré J, Prévotat A, Tardif C, Langlois C, Duhamel A, Wallaert B. Determinants of exercise capacity in cystic fibrosis patients with mild-to-moderate lung disease. BMC Pulm Med. 2014;14:74.
Article PubMed PubMed Central Google Scholar
Pianosi P, Pelech A. Stroke volume during exercise in cystic fibrosis. Am J Respir Crit Care Med. 1996;153(3):1105–9.
Szollosi I, King SJ, Wilson JW, Naughton MT. Tachycardia in adults with cystic fibrosis is associated with normal autonomic function. Intern Med J. 2011;41(6):455–61.
Hebestreit H, Hulzebos EHJ, Schneiderman JE, Karila C, Boas SR, Kriemler S, et al. Cardiopulmonary exercise testing provides additional prognostic information in cystic fibrosis. Am J Respir Crit Care Med. 2019;199(8):987–95.
Article PubMed Google Scholar
Edgeworth D, Keating D, Ellis M, Button B, Williams E, Clark D, et al. Improvement in exercise duration, lung function and well-being in G551D-cystic fibrosis patients: a double-blind, placebo-controlled, randomized, cross-over study with ivacaftor treatment. Clin Sci (Lond). 2017;131(15):2037–45.
Wilson J, You X, Ellis M, Urquhart DS, Jha L, Duncan M, et al. VO2max as an exercise tolerance endpoint in people with cystic fibrosis: lessons from a lumacaftor/ivacaftor trial. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2021;20(3):499–505.
Article CAS Google Scholar
Saynor ZL, Barker AR, Oades PJ, Williams CA. The effect of ivacaftor in adolescents with cystic fibrosis (G551D mutation): an exercise physiology perspective. Pediatr Phys Ther Off Publ Sect Pediatr Am Phys Ther Assoc. 2014;26(4):454–61.
Google Scholar
Savi D, Schiavetto S, Simmonds NJ, Righelli D, Palange P. Effects of Lumacaftor/Ivacaftor on physical activity and exercise tolerance in three adults with cystic fibrosis. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2019;18(3):420–4.
Ahmed MI, Dayman N, Madge J, Gaillard E. P91 Cardiopulmonary exercise testing in CF adolescents after starting Tezacaftor/Ivacaftor. Thorax. 2021;76(Suppl 1):A136–A136.
Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–19.
Article CAS PubMed PubMed Central Google Scholar
Snowball JE, Flight WG, Heath L, Koutoukidis DA. A paradigm shift in cystic fibrosis nutritional care: clinicians’ views on the management of patients with overweight and obesity. J Cyst Fibros. 2023;0(0). https://doi.org/10.1016/j.jcf.2023.03.011 .
Causer AJ, Shute JK, Cummings MH, Shepherd AI, Wallbanks SR, Pulsford RM, et al. Elexacaftor-Tezacaftor-Ivacaftor improves exercise capacity in adolescents with cystic fibrosis. Pediatr Pulmonol. 2022;57(11):2652–8.
Hebestreit H, Arets HGM, Aurora P, Boas S, Cerny F, Hulzebos EHJ, et al. Statement on exercise testing in cystic fibrosis. Respir Int Rev Thorac Dis. 2015;90(4):332–51.
Rysgaard UK, Pedersen CL, Jensen JH, Sørensen L, Philipsen LKD, Leo-Hansen C, et al. Change in exercise capacity measured by Cardio-pulmonary Exercise Testing (CPET) in Danish people with cystic fibrosis after initiation of treatment with Lumacaftor/Ivacaftor and Tezacaftor/Ivacaftor. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2022;21(5):844–9.
Caterini JE, Ratjen F, Barker AR, Williams CA, Rendall K, Schneiderman JE, et al. Exercise intolerance in cystic fibrosis-the role of CFTR modulator therapies. J Cyst Fibros Off J Eur Cyst Fibros Soc. 2022;21(2):282–92.
Warth JD, Collier ML, Hart P, Geary Y, Gelband CH, Chapman T, et al. CFTR chloride channels in human and simian heart. Cardiovasc Res. 1996;31(4):615–24.
Robert R, Norez C, Becq F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl- transport of mouse aortic smooth muscle cells. J Physiol. 2005;568(Pt 2):483–95.
Lamhonwah AM, Bear CE, Huan LJ, Kim Chiaw P, Ackerley CA, Tein I. Cystic fibrosis transmembrane conductance regulator in human muscle: Dysfunction causes abnormal metabolic recovery in exercise. Ann Neurol. 2010;67(6):802–8.
Malik FA, Meissner A, Semenkov I, Molinski S, Pasyk S, Ahmadi S, et al. Sphingosine-1-phosphate is a novel regulator of cystic fibrosis transmembrane conductance regulator (CFTR) activity. PLoS One . 2015;10(6):e0130313.
Madácsy T, Pallagi P, Maleth J. Cystic fibrosis of the pancreas: the role of CFTR channel in the regulation of intracellular Ca2+ signaling and mitochondrial function in the exocrine pancreas. Front Physiol. 2018;9:1585.
Velsor LW, Kariya C, Kachadourian R, Day BJ. Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Respir Cell Mol Biol. 2006;35(5):579–86.
van de Weert-van Leeuwen PB, Slieker MG, Hulzebos HJ, Kruitwagen CLJJ, van der Ent CK, Arets HGM. Chronic infection and inflammation affect exercise capacity in cystic fibrosis. Eur Respir J. 2012;39(4):893–8.
Casey M, Gabillard-Lefort C, McElvaney OF, McElvaney OJ, Carroll T, Heeney RC, et al. Effect of elexacaftor/tezacaftor/ivacaftor on airway and systemic inflammation in cystic fibrosis. Thorax. 2023;78(8):835–9.
Divangahi M, Balghi H, Danialou G, Comtois AS, Demoule A, Ernest S, et al. Lack of CFTR in skeletal muscle predisposes to muscle wasting and diaphragm muscle pump failure in cystic fibrosis mice. PLoS Genet. 2009;5(7):e1000586.
Download references
Acknowledgements
We appreciate the willingness of all patients and medical staff to participate in clinical trials and their processing.
Publication will be supported by the Czech Pulmonological and Phthisiological Society (open access publication free grant).
Author information
Authors and affiliations.
Department of Pulmonology, Second Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czech Republic
Nela Stastna & Libor Fila
Faculty of Medicine, Masaryk University, Brno, Czech Republic
Nela Stastna
Department of Sports Medicine and Rehabilitation, St. Anne’s University Hospital and Faculty of Medicine, Masaryk University, Brno, Czech Republic
Lenka Hrabovska & Pavel Homolka
Department of Paediatric Infectious Diseases, University Hospital Brno and Faculty of Medicine, Masaryk University, Brno, Czech Republic
Lukas Homola
Institute of Biostatistics and Analyses Ltd. and Faculty of Medicine, Masaryk University, Brno, Czech Republic
Michal Svoboda
Department of Pulmonology, University Hospital Brno and Faculty of Medicine, Masaryk University, Brno, Czech Republic
Kristian Brat
You can also search for this author in PubMed Google Scholar
Contributions
N.S.: writing – original draft, methodology, visualization, formal analysis, data curation, L.H.: supervision, methodology, P.H.: writing – original draft, methodology, formal analysis, data curation, L.H.: methodology data curation, M.S.: data curation, formal analysis, K.B.: supervision, writing – review, editing, L.F.: supervision, writing – review, editing.
Corresponding author
Correspondence to Nela Stastna .
Ethics declarations
Ethics approval and consent to participate.
Approved by University Hospital Brno committee.
Informed consent was obtained from all subject or their legal guardians.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Stastna, N., Hrabovska, L., Homolka, P. et al. The long-term effect of elexacaftor/tezacaftor/ivacaftor on cardiorespiratory fitness in adolescent patients with cystic fibrosis: a pilot observational study. BMC Pulm Med 24 , 260 (2024). https://doi.org/10.1186/s12890-024-03069-8
Download citation
Received : 19 March 2024
Accepted : 21 May 2024
Published : 28 May 2024
DOI : https://doi.org/10.1186/s12890-024-03069-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
BMC Pulmonary Medicine
ISSN: 1471-2466
- Submission enquiries: [email protected]
- General enquiries: [email protected]
COMMENTS
Case Study - Cystic Fibrosis (Remote Edition) KEY 1. "Woe to that child which when kissed on the forehead tastes salty. He is bewitched and soon will die" This is an old saying from the eighteenth century and describes one of the symptoms of CF (salty skin). Why do you think babies in the modern age
Case study 1: Cystic fibrosis. cystic fibrosis. Click the card to flip 👆. A genetic disorder that occurs in people with two copies of a certain recessive allele; characterized by an excessive secretion of mucus and consequent vulnerability to infection; fatal if untreated. Click the card to flip 👆.
Study with Quizlet and memorize flashcards containing terms like Which statement by the mother supports the diagnosis of CF?, Which documentation further supports the diagnosis of CF? A history of frequent respiratory infections. An elevated white blood cell count (WBC). Reports of episodic abdominal pain and crying. A sweat chloride level of 35 mEq/L. Reports of foul-smelling stools that have ...
suggest that they add extra salt to debbie's diet and watch her for dehydation. Study with Quizlet and memorize flashcards containing terms like Which statement by the mother supports the diagnosis of CF, which documentation further supports the diagnosis of CF, what information will the nurse include when teaching about the sweat test and more.
This case study is a follow-up to the Cystic Fibrosis Case Study where students explore how changes in transport proteins affects the movement of ions, resulting in a build-up of chloride ions and the symptoms of the disease.. Students were introduced to the idea that different mutations can cause differences in the transport proteins, but in the first version, the origin of these mutations ...
3. What is the probability that Joshua's brother has cystic fibrosis? Because both parents are heterozygous (Nn), there is a 25% chance of each child inheriting two "n" alleles and having cystic fibrosis. Case Study #2 Answers 1. Would you predict that the skin condition is most likely inherited from a recessive allele?
Cystic fibrosis (CF) is an autosomal recessive condition affecting approximately 30,000 Americans and 70,000 people worldwide.According to the Cystic Fibrosis Foundation (Cystic Fibrosis Foundation, 2019a), approximately 1,000 new cases are diagnosed yearly in the United States, with a known incidence of 1 per 3,900 live births.The disease prevalence varies greatly by ethnicity, with the ...
View Cystic Fibrosis CS Answer Sheet (1).docx from BIOL 101 at North Carolina A&T State University. Case Study : Cystic Fibrosis Case Study Answer Sheet Type your answers in red Type your answers in
This case introduces students to "Maggie," who has just been diagnosed with cystic fibrosis. The students must identify the mutation causing Maggie's disease by transcribing and translating a portion of the wildtype and mutated CFTR gene. Students then compare the three-dimensional structures of the resulting proteins to better understand ...
Mix the pancreatic enzymes with hot, starchy foods such as macaroni or pasta. Open the enzyme capsules and mix the beads in a protein food. Ensure that the child swallows the pancreatic enzyme capsule whole. Ensure that enzymes are administered within 30 min of consuming meals and snacks.
Case Study: Cystic Fibrosis and Mutations Answer KEY Student Version: AI Homework Help. Expert Help ... Total views 100+ Grand Canyon University. MATH. MATH HLT3624. colly678. 2/16/2022. 100% (1) View full document. Students also studied. Cystic Fibrosis CS Answer Sheet (1).docx. Solutions Available. North Carolina A&T State University. BIOL ...
Part I: A Case of Cystic Fibrosis. Dr. Weyland examined a six month old infant that had been admitted to University Hospital earlier in the day. The baby's parents had brought young Zoey to the emergency room because she had been suffering from a chronic cough. In addition, they said that Zoey sometimes would "wheeze" a lot more than they ...
View Homework Help - Case Study 15 - Cystic Fibrosis from HCR 240 at Mesa Community College. Case Study 15 - Cystic Fibrosis 1) Which of the following best explains why the patient in this case study ... Both parents are carriers of a mutation for cystic fibrosis. This answer made sense to me because the case study does mention that the mother ...
of school, doesn't have any energy, is coughing more, and is "having a hard time breathing." 1. What additional data should be obtained from JR and his parents? of school, doesn't have any energy, is coughing more, and is "having a hard time breathing." ... Where do you study. Your language. The Netherlands. United Kingdom. Germany ...
Elfeky M, Cystic fibrosis. Case study, Radiopaedia.org (Accessed on 29 May 2024) https://radiopaedia.org/cases/179364
Case Study: During late spring Erin's breathing has become increasing congested over the past week and her parents suspect that she has developed a respiratory infection when she becomes febrile with a temperature of 37.9 0 C (100.2 0 F).
Her youngest child was diagnosed with cystic fibrosis while in the NICU. Jennifer has always been passionate about writing, majoring in English in college and teaching a creative writing workshop to middle school students upon graduation. She went on to receive her law degree and is currently a practicing attorney.
Study with Quizlet and memorize flashcards containing terms like The nurse suspects that Darla may have Cystic Fibrosis (CF). Which statement by the mother supports the diagnosis of CF? A. My daughter went to bed feeling fine last night and woke up with a high fever and a sore throat B. Lately Darla's eyes appear to have a yellow discoloration C. When i kiss my daughter her skin tastes like ...
Ultimately, the research may have medical applications, though ironically not likely for most cystic fibrosis patients. Because two-thirds of cystic fibrosis patients fail to produce the cystic fibrosis channel altogether, a cure for most is expected to result from research focused on replacing the lost channel. 6.
A new study hopes to find out whether cystic fibrosis could be treated in utero . ... Bringing answers to children with a little-known genetic condition. Read more. But as well as bringing joy, this creates a challenge. Because CF treatments are currently only approved for children over two years old, people are seeking much-needed guidance on ...
Case Study Cystic Fibrosis Answers 1. Increased viscosity of mucus glands secretions leads to increased viscosity of bronchial mucus producing greater resistance to ciliary action, slower flow of mucus, incomplete expectoration which contributes to mucus obstruction. Retained mucus serves as a growth medium for bacteria growth, leading to destruction of lung tissue.
Abstract Cystic fibrosis (CF) is a rare autosomal recessive genetic disorder that is now commonly treated with cystic fibrosis transmembrane conductance regulator (CFTR) modulators. ... We present a case report of a young adult with rapid CFTR-modulator-induced worsening of pre-existing acne vulgaris refractory to minocycline treatment, which ...
Background. Physical activity is a crucial demand on cystic fibrosis treatment management. The highest value of oxygen uptake (VO 2peak) is an appropriate tool to evaluate the physical activity in these patients.However, there are several other valuable CPET parameters describing exercise tolerance (W peak, VO 2VT1, VO 2VT2, VO 2 /HR peak, etc.), and helping to better understand the effect of ...
426 terms. emmaacox8. Preview. peds 3. 16 terms. Annabella_Tayamen. Preview. Study with Quizlet and memorize flashcards containing terms like Cystic Fibrosis is ..., Signs and Symptoms of Cystic Fibrosis, Most common test for Cystic Fibrosis and more.
According to the Cystic Fibrosis Foundation, Cystic fibrosis is an inherited chronic disease that affects the lungs and digestive system of about 30,000 children and adults in the United States (70,000 worldwide). A defective gene and its protein product cause the body to produce unusually thick, sticky mucus that:
Week 6 Lecture Quizzes: Protein Synthesis (case: Cystic Fibrosis) the CFTR protein is a growth factor receptor four don the surface of lung cells. Click the card to flip 👆. False; the CFTR protein is a membrane protein responsible for controlling the movement of chloride in and out of cells. Click the card to flip 👆.
DOI: 10.1016/j.transproceed.2024.03.034 Corpus ID: 269785319; Combined Liver-Pancreas Transplantation as Novel Treatment for Patient With Cystic Fibrosis: A Case Report. @article{Zienkiewicz2024CombinedLT, title={Combined Liver-Pancreas Transplantation as Novel Treatment for Patient With Cystic Fibrosis: A Case Report.}, author={Damian Zienkiewicz and Paulina Kalman and Pawel Skrzypek and ...