Annotating Digitally
- Which Tool Is Right For Me?
- Use Adobe Reader
- Use Hypothesis
- Use Preview App for Mac
- Microsoft Edge for Windows 10

Annotating with Hypothesis
Hypothesis is a free online tool designed to allow for collaborative annotation across the web. This guide will walk you through using Hypothesis with the Chrome extension. To use the bookmarklet in another browser, please refer to the hypothesis user guide for step-by-step instructions.
It can be used to annotate web pages, PDFs and EPUB files. You can annotate documents and pages publicly, privately, or within a group.
To get started with Hypothesis you'll need to:
- Create an account.
- Install the Chrome extension or the Hypothesis Bookmarklet if you are using a browser other than Chrome.
- Start annotating!
What makes Hypothesis different than other tools is that you can annotate PDFs as well as web pages. It also allows for group and collaborative annotation.
- Create a Hypothesis Account
- Chrome Extension
- Hypothesis User Guide
Opening Web Pages and PDFs
Navigate to the web page or pdf in your browser.
Next, select the Hypothesis icon located in the right corner of your Chrome browser if it is not already active.

An inactive icon will appear as light gray, and if it is ready to use it will be black. If you have not used Hypothesis for a while you may need to log in to activate the program.
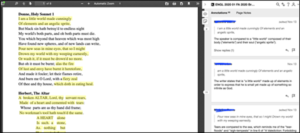
Once Hypothesis is active on a page you can start annotating. You can highlight text, add annotation, add a note for the whole page, share the page with others, and reply to other comments on the page. You may have to click the toggle arrow to see options, comments, and notes.

Adding Annotation and Highlighting Text
To highlight or add annotation for certain text, simply select the text and the highlight and annotation icons will appear.

To create a note for the entire page, select the note icon that appears on the Hypothesis menu on the right side of the screen. You can also toggle highlighted text on and off using the eye icon.

Formatting Annotation
Once you have selected text to annotate, an annotation box will appear in the menu on the right. The annotation section allows you to perform minor formatting of text such as bold, italics, quoted text, lists, links, and even mathematical notation that is LaTex supported.

There is also an option to add web links and links to online images and videos. Images will appear and videos will play right in the annotation section of the page.

Adding Tags
You may also add tags to your annotation to better organize your idea and thoughts, and categorize content. To learn more about tags and how they can be used to enhance collaboration and search, please see the Using Tags Tutorial .

Public, Private, and Private Group Annotation
Annotations, notes and highlights can be made public, private or shared within a specific group. All annotation setting default to public unless you choose another option. Public annotations can be seen by anyone who visits the webpage or pdf and has Hypothesis enabled.
Switching from Public to "Only Me"
If you would like to make notes private where only you can see them, be sure to select the Only Me option in the annotation box.

Once you have selected this option, all other annotations, highlights and notes will default to private on the page and be shown with a lock icon next to your name. To switch back select the Public option.

Private Groups
To share annotations with a group you have already created or have been invited to, select the down arrow next to Public in the Hypothesis menu. Here you will see any private groups you belong to as well as an option to create a new private group. Selecting a group will allow you to annotate and have only the members belonging to the group view and respond to your comments, highlights and notes.

To create a new group, select the + New Private Group option. From here you will be taken to a new screen where you will be asked to name the group. Click the Create group button to create the new private group.

Once the group is created, you will be given a link to share to invite new members to the group.

Annotating a Locally-Saved PDF
To annotate a saved pdf in Hypothesis, open it in your browser. Once it is open in your browser activate Hypothesis to start annotating. The pdf must have selectable text in order for Hypothesis to work. If you have issues with this feature, consult the annotating locally saved pdfs tutorial .

- << Previous: Use Adobe Reader
- Next: Use Preview App for Mac >>
- Last Updated: Apr 17, 2024 10:22 AM
- URL: https://libguides.trinity.edu/digital-annotation
- Request a Consultation
- Workshops and Virtual Conversations
- Technical Support
- Course Design and Preparation
- Observation & Feedback
Teaching Resources
Resource Overview
Guide on using Hypothesis, a social annotation tool used for collaboration, interactive reading, annotation, and discussion.
For more ideas and technical support, email the EdTeam team or schedule a consult . You can also reach out to Becky George, WashU Customer Support Specialist at Hypothes.is, to get individual help and ideas for using Hypothes.is by emailing her or scheduling a consult with her.
- WashU licensed
- Seamlessly integrates with Canvas Assignments, Groups, and SpeedGrader
- Shared annotations and replies as well as private highlights and notes
- Use with webpages or searchable PDFs
- Can split students into small reading groups
- Ability to annotate with images, links, and videos
- Student resources, including instructions , tutorials , and guides on best practices and annotation types
- CTL managed and supported
Hypothesis makes reading active, visible, and social. A metanalysis conducted by Novak et al in 2012 suggested that social annotation tools can “lead to learning gains in higher education” (p. 47). In particular, the study found that social annotation activities contribute to “improved critical thinking, meta-cognitive skills, and reading comprehension” (p. 47). Preliminary findings in the same study also suggested that using social annotation “promotes motivation for reading and contributes to higher frequency of positive emotions and lower frequency of negative emotions” (towards the reading). In addition, Brown and Croft (2020) argue that social annotation is an inclusive teaching practice because of how it promotes collaborative knowledge building.
- Increase engagement on a reading assignment by asking students to share reactions, personal connections, or questions about a text. Such annotations make the reading experience social and spark conversations between students.
- Familiarize students with the expectations and objectives of your course by having them annotate the syllabus. This not only ensures that students read the syllabus, but also helps build community from the start .
- Help students read a difficult text together. In the instructions, ask students to raise questions about confusing concepts, share how they interpret the text, and add links to resources that define terms, provide examples or elaborate on concepts raised in the text.
- Help students do more close reading by asking probing questions or directing students to important parts of the text. Open the text in Hypothesis as a student would and add your prompts as annotations. Consider these 10 different types of annotations .
- Use a text or a section of a text to model how you would engage with it. Open the text in Hypothesis and add annotations to share your own responses, analyses, and meta-comments. You could also use this opportunity to introduce students to annotation etiquette , or best practices that make annotations more useful for others.
- Synthesize annotations using tags. Make sure that you and students add specific tags to your annotations such as by concept, topic, or comment type (e.g., “metaphor” “love” “resource” “follow-up”). This will allow you to search for this tag and quickly identify themes across annotations.
- Include multimedia in annotation by showing students how to add external images and videos in comments and replies. These can be immediately viewed in the comments window.
- Get ideas from the Hypothesis teacher community at Liquid Margins which meets regularly to discuss ideas for social annotation. All past recordings are available on YouTube .
- Discussion Board Alternative: Students benefit from conversations that take place within a text. This can keep their conversations specific and grounded. Students can post questions and then respond to other students’ questions OR you can place questions within the text for students to respond to.
- Comparative Activity: Students work to closely analyze a series of texts looking for commonalities, dissimilarities, or other points of importance for future discussion.
- Close Reading Practice: Students often fly through short complex texts too quickly. A Hypothesis activity can slow them down and encourage them to engage in slow, careful reading. Students may be tasked with adding definitions to unfamiliar words, grappling with passages that seem critical or patterns that stand out in the text, and linking to other texts or theories that a passage seems to allude to etc.
- Rhetorical Analysis: Students might be tasked with marking and explaining rhetorical strategies that they encounter in a text.
- Multimedia Writing: Students use Hypothesis to create a collaborative multimedia text by annotating a written text with images and video.
- Creative Writing Exercise: Students could respond creatively and collaboratively to a written text. It could also be used as an exercise (e.g. annotate “x” text in the voices of characters from a prior text).
- Syllabus Search : Students annotate for specific important course details such as due dates, course goals, and resources. This is a great beginning activity to build community and help students identify how your course relates to students’ personal and professional goals.
- Jigsaw Multiple Texts : Students are split into separate groups where each group reads a different text. After becoming “experts” on their assigned reading, they split into new groups with experts of other texts to teach each other about the text that they read.
- Brown, M. & Croft, B. (2020). “Social annotation an inclusive praxis for open pedagogy in the college classroom.” Journal of Interactive Media in Education, 1-8.
- Novak, E. et al. (2012). “The educational use of social annotation tools in higher education: A literature review.” The Internet and Higher Education, 15, 39-49.
Get Started
Hypothesis is fully integrated into Canvas. You’ll find it under Assignments and External Tools.
Here’s a great page designed to walk faculty through social annotation and using Hypothesis: https://web.hypothes.is/getting-started-with-canvas/
Hypothesis Tutorials
- How to set up and grade a Hypothesis Activity
- Quick 2-page overview
Additional Tutorials from Hypothesis (external links)
- How to set up Hypothesis readings through Canvas Assignments
- How to set up Hypothesis readings through Canvas Modules
- How to set up Hypothesis reading groups in Canvas
- How to grade Hypothesis annotations in Canvas
Ideas for How to Use Hypothesis
- Starter assignment ideas
- User-submitted assignment ideas
- NEW! AI Starter Assignments with ChatGPT
- Instructions for Students
- A student guide to Hypothesis in Canvas
- An Illustrated Guide to Annotation Types
- Annotation tips for students
- How to sync Hypothesis readings for imported Canvas courses
Have suggestions?
If you have suggestions of resources we might add to these pages, please contact us:
[email protected] (314) 935-6810 Mon - Fri, 8:30 a.m. - 5:00 p.m.
- The Student Experience
- Financial Aid
- Degree Finder
- Undergraduate Arts & Sciences
- Departments and Programs
- Research, Scholarship & Creativity
- Centers & Institutes
- Geisel School of Medicine
- Guarini School of Graduate & Advanced Studies
- Thayer School of Engineering
- Tuck School of Business
Campus Life
- Diversity & Inclusion
- Athletics & Recreation
- Student Groups & Activities
- Residential Life
Teach Remotely
How to Teach From Anywhere
Hypothesis Annotation Guide
- Introduction
The Hypothesis annotation tool is now available in all Dartmouth Canvas sites. Hypothesis brings discussion directly to course content by enabling students and teachers to add comments and start conversations in the margins of instructional texts and other resources. Collaborative annotation creates an opportunity for students to engage more deeply in course readings and gives teachers a view of how students are reading.
Hypothesis also connects directly to your Canvas site and allows students to annotate documents you provide through Canvas Files.
- Course Activity Ideas
Are you looking for ideas about how to use collaborative annotation in your course activities? The following are activities that could pair well with this approach:
- Peer feedback and editing - similar to the Peer Review tool in Canvas, you can create peer review activities and group annotations.
- Qualitative coding - for methods courses, consider bringing in example data for qualitative coding where students can practice and get feedback on developing skills.
- Close reading and primary source analysis - Consider having students practice close reading and analysis skills with source material in a collaborative context.
- “Debugging” activities - Collaborative annotation can allow students to help peers to “debug” code and other development projects.
- Case study analysis - Courses working with case studies could use collaborative annotation as a way to surface key information from case materials. It also allows the instructor to see into the student preparation process for class.
- Interpreting information, data, visuals, or artifacts - Numerous courses incorporate practicing critical reading and interpretation with a variety of materials. Collaborative annotation offers an opportunity to surface details around those practices.
Getting Started and Using Hypothesis in Canvas
Hypothesis, is accessed through Canvas Assignments [as an External] tool for both graded or ungraded activities. To get started and learn more see the following guides:
Getting started with Hypothesis:
- Annotation tips and etiquette for students
- An Illustrated Guide to Annotation Types
- Using images, links, and videos in annotations
Using Hypothesis in Canvas:
- How to set up Hypothesis readings through Canvas Modules
- How to set up Hypothesis readings through Canvas Assignments
- How to grade Hypothesis annotations in Canvas
- A student guide to Hypothesis in Canvas
Dartmouth Hypothesis Workshop (March 2020) slides
For questions or support using Hypothesis faculty can contact, [email protected] or for questions about connection to your Canvas site or assignment design contact, [email protected]
Hypothesis Guide
- Getting Started
Subscribe By Email
Your Email Leave this field blank
This form is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.

Pressbooks 101
You might be familiar with Hypothes.is as a web annotation tool that you use through a browser extension, or by pasting a link in on their website. Now, you can harness the annotation power of Hypothes.is in Pressbooks. The annotator now comes built into core Pressbooks. You can use Hypothes.is as an annotator for editing groups working on a book, to handle peer review, and even as a classroom tool for students.
In this chapter, you’ll learn how to:
- Activate Hypothes.is for your Pressbook
- Register for a Hypothes.is account
- Create annotation groups
- Review annotation activity
- Access additional Hypothes.is resources
Activate Hypothes.is for your Pressbook
By default Hypothes.is is not activated in your Pressbook. In order to turn on the annotation tool, click Settings on the right-hand menu, then click Hypothesis. This will load the Hypothesis Settings Page.
The Hypothesis Settings menu includes:
- Highlights on by default – check this box if you want the annotation highlights to be turned on by default for your book.
- Sidebar open by default – check this box if you want the annotation pane to be open by default on your book.
- Enable annotation for PDFs in Media Library – check this box if you want to enable annotation for PDFs in your media library.
- Content Settings – these check boxes control where Hypothes.is is loaded on your book. They include the front page, blog page, parts, chapters, front matter, and back matter.
- Allow – these fields allow you to apply Hypothes.is to specific parts, chapters, front matter, and back matter.
- Disallow – these fields allow you to turn off Hypothes.is for select specific parts, chapters, front matter, and back matter.

Register for a Hypothes.is Account
To use Hypothes.is you’ll need to register for an account . To do so you’ll need to provide a username, email address, and password, and then click the Sign Up button. After that, you should receive a confirmation email with a link to click to validate your account. If you don’t see this email check your Spam or Junk folder. [1]
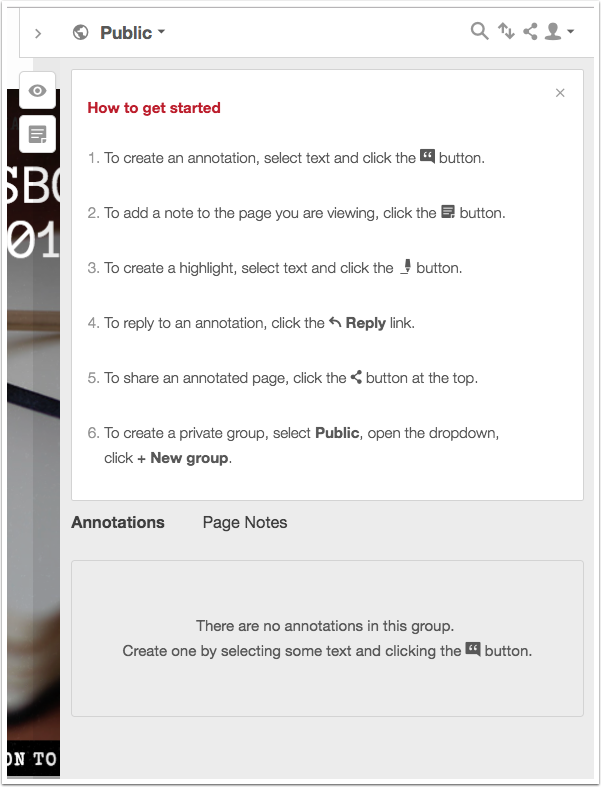
The first time you log into your Hypothes.is pane the How to Get Started box is displayed. It offers explanations of the different button functions and briefly how to use them.
To create an annotation or highlight, start by selecting the text and then choose either annotate or highlight. Choosing annotation will open the annotation pane to a new blank annotation field. Selecting highlight will highlight the text you have selected.

Annotation Pane Tour
- Filter, Sort, Share, and Account buttons
- Turn on/off highlights, create annotation buttons
- Text editor formatting tools – bold, italic, quotation, link, media, LaTex, numbered list, bulleted list,
- Preview annotation
- Tags Field – add multiple tags separated by commas
- Post button – you can use the drop-down button to easily switch between Public, Private, and Group posting

Once you’ve created an annotation it will appear in the annotation pane on the right side of the page. It features the text that you’ve selected to annotation at the top, your annotation, and tags.
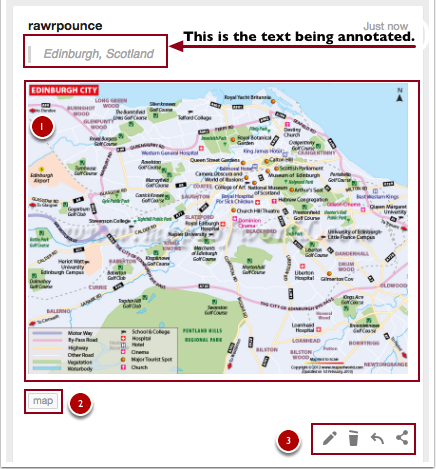
In the screenshot below, draw your attention to (1) the annotation, in this case, I’ve provided a map of Edinburgh, Scotland to show other users what the city’s layout looks like, (2) what tags look like when you use them on an annotation, (3) the edit, delete, reply, and share buttons.
You can also adjust the width of the annotation pane by dragging it to left (wider) or to the right (narrower).
Creating a Group
I recommend following the Hypothes.is Creating a Group step-by-step guide video tutorial. While it displays annotation on a web page, it is accessed through the Hypothes.is panel the same way it would be on a Pressbook.
Joining a Group
I recommend the following Hypothes.is Joining a Group step-by-step guide video tutorial. This might be helpful to show students if you’re using a group to manage class annotations.
Review Annotation Activity
Once you’re the member of a group, you can easily review the annotation activity for that group.
To navigate to the annotation activity for a specific group, open the annotation pane on your book and select the user icon to open the drop-down tray of options, then click on your username.
This will load your account page, from there click Groups and from the drop-down tray select the group you want to review. For example, in the screenshot, the PPP Editors group is selected.
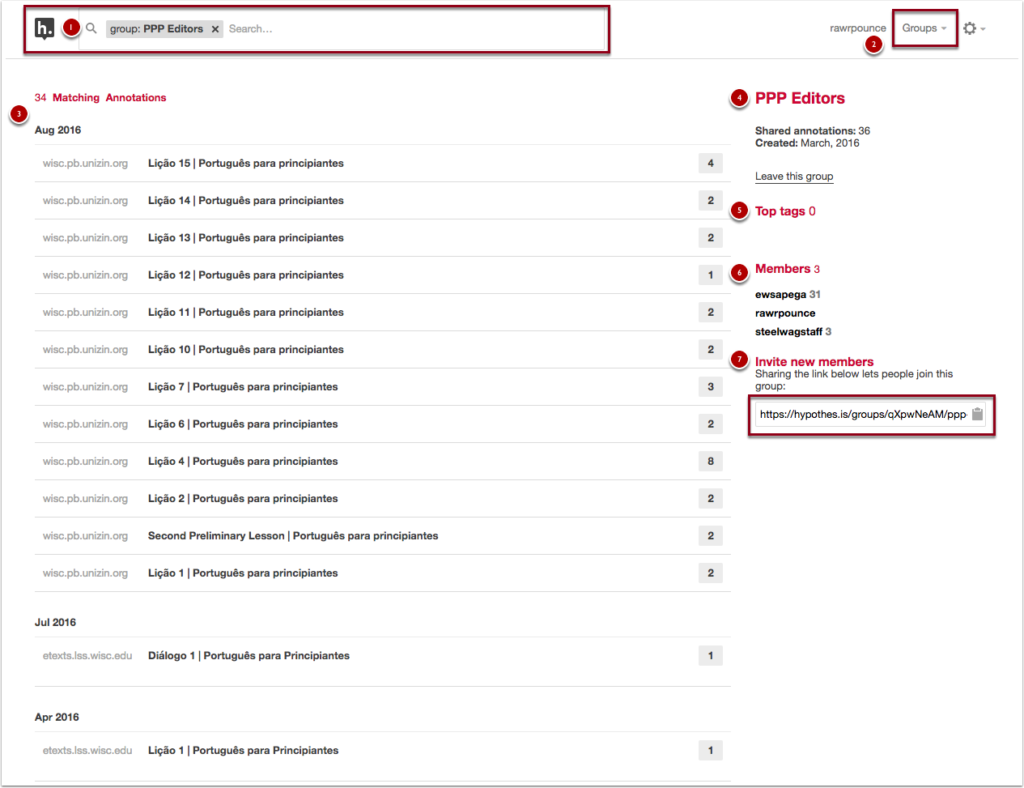
This will load the group page that you’ve selected. At the top of the page you’ll find (1) the group you’ve selected as a filter in the search box, (2) the account and group navigation options in the top right of the page, (3) a listing of the matching annotations for the group tag, (4) the name of the group and group information, (5) a listing of the top tags for the group, (6) a list of members in the group, and (7) the link to invite new members to the group.
Hypothes.is offers some great resources for using annotations with students and in groups. Check out the following resources:
Back to School with Annotation: 10 Ways to Annotate with Students – offers 10 creative ways to use annotation with your students.
Annotation Tips for Students – offers tips aimed towards helping students complete annotations.
Hypothes.is Education – offers more guides and classroom examples for using Hypothes.is.
You can also check out the Hypothes.is YouTube channel tutorials .
- In the future, we hope to have a single sign-on using Wisc NetId. ↵
Pressbooks 101 Copyright © 2017 by L & S Learning Support Services is licensed under a Creative Commons Attribution 4.0 International License , except where otherwise noted.
Web Request Blocked
Your request has been identified as a security risk and has been blocked by TeamDynamix. If you believe the request is valid, please report the blocked web request. You'll need to include the Blocked Request Url and Support ID in your report.
Center for Teaching Innovation
Hypothesis is a social annotation tool that quickly allows you to make class readings more active, visible and social. For more information on social annotation, see Annotation 101 and Back to School with Annotation: 10 Ways to Annotate with Students
Annotation can help students with reading comprehension and developing critical thinking about course materials. Hypothesis allows students to annotate online readings. Digital annotation also offers new possibilities such as responding to text using different media and empowers students to collaborate on understanding and developing ideas about their readings.
Cornell faculty and students can access Hypothesis via Canvas, where it is found as an external tool in either assignments or modules.
Considerations for using Hypothesis
Hypothesis is built on an open-source platform and has a strong and stated position on learner privacy. While it has fewer bells and whistles than Perusall, its emphasis on user experience, clean interface, and ease of use makes it a strong choice.
Hypothesis can help you create a searchable PDF, or if you prefer to do this in advance, the Cornell library provides information on methods for creating searchable PDFs .
More Information
- Annotation 101
- Ten Ways to Annotate with your Class
Hypothesis: Collaborative Annotation for Canvas LMS
Available for.
Available for : Instructors Students
Log in to Canvas to access Hypothesis
ABOUT HYPOTHESIS
Hypothesis is a collaborative annotation tool integrated with Canvas that supports shared annotations within a course, discussion in response to annotations, and active reading of text. Instructors select Hypothesis as an external tool when setting up an assignment and can also choose to assign readings to groups. Students can then annotate course readings collaboratively, sharing comments, and replying to each other’s comments with text, links, images, and video. Hypothesis is also fully integrated with SpeedGrader for efficient review and grading of student annotations.
Resources for instructors
- We recommend selecting the Load In A New Tab option when setting up a Hypothesis assignment. This will allow for a better reading experience for students, especially those who magnify the contents of their screen for accessibility purposes.
- Set up Hypothesis readings through Canvas Modules
- Grade Hypothesis annotations in Canvas
- If you are using Canvas Files or Groups for any Hypothesis readings you will need to take additional steps before the assignment works in the new course.
- Hypothesis FAQs
Resources for students
Consider sharing the following links in your Canvas course, or point students to this page
- Learn the basics of navigating and using Hypothesis
- Short screen casts show how to highlight, annotate, make page notes, and reply to others’ notes
- Jazz up your annotations with this deep dive into the editing interface
- Create stand-out annotations with these five best practices to make your annotations stand out
Hypothesis helps you to
- Provide a new way for students to discuss class readings
- Help students consider multiple viewpoints when reading
- Assist students in close and active reading of texts
- Encourage students to engage critically with readings
Hypothesis Support
Workshops & webinars, hypothesis 101.
If you’d like to learn more about Hypothesis and see a demo, register for an upcoming Hypothesis 101 webinar or watch a Hypothesis 101 recording .
Hypothesis Partner Workshops
Each quarter, Hypothesis offers a variety of (typically) 30min workshops led by their team . Are you looking for ways to help your students develop their close reading skills and increase their engagement with your course materials? Maybe you’re seeking a more collaborative approach to reading complex texts while building community? Get ideas you can bring back to your courses, students, and colleagues for how to use Hypothesis for social annotation.
Topics for this quarter:
- Activating annotation in Canvas
- Using multimedia & tags in annotations
- Using Hypothesis with small groups
- Creative ways to use social annotation in your course
- Show-and-tell participatory workshop
Liquid Margins
Hypothesis hosts a recurring web “show” featuring instructors and staff to talk about collaborative annotation, social learning, and other ways to make knowledge together.
Offered throughout the year
Previous workshop recordings
If you missed any of the Hypothesis partner workshops offered during autumn quarter, you can find recordings on the Hypothesis YouTube channel .
Vendor Help
- The Hypothesis Knowledge Base includes FAQs, tutorials, how-tos, and troubleshooting tips.
- Schedule a meeting with Hypothesis Customer Success Specialist Autumn Ottenad for instructional design advice or questions on how to best use Hypothesis in your course.
- Watch Liquid Margins , the Hypothesis web series, to learn more about how other instructors use collaborative annotations in their course.
- Email and phone

Pedagogy Playground
Innovative Teaching in Higher Ed

Annotating Readings with Hypothesis
In my classes, I like employing strategies that promote deep reading. I started using Hypothesis in one of my graduate classes this semester to help students connect with concrete quotes from the text while also analyzing and reflecting on them. This system has the additional benefit of holding students accountable to engaging with the assigned readings. Annotating with hypothesis also offers a social component, one that extends the discussion of course materials outside of the classroom. Mia C. Zamora , an Associate Professor of English and Director of the Kean University Writing Project, explains that tools like Hypothesis “extend the discursive space of the classroom” which “facilitates engaged reading practices.” She continues, “A collaborative reading environment seems to me a fair foundation for dynamic and thoughtful interaction, which, in turn, holds the potential to mirror the kind of rich and complex dialogue we aspire to in a working democracy.” So far, I’ve found this tool to promote thoughtful student engagement with class texts while also sharing thoughts and ideas with each other and with me prior to the start of class each week.
Basics of the Tool
- You can use Hypothesis in a browser — either as a Chrome plug in or via a link “preface” (a snippet of a link that you add onto the web address you want to annotate). The browser version works well for readings that are websites, blog posts, and PDFs online. There’s also an LMS version that you can add to Canvas, Blackboard, and other LMS systems. If you have a lot of scanned PDFs that are not available freely online, this is a good route to take. A different tool, Perusall , allows students to annotate e-books/e-textbooks, although the selection of texts is limited.
- Hypothesis allows editing in public, in private groups, or privately . I use a private group to create a class-specific space where we can engage with each other without having to filter out comments left by others. A private group also provides a space with greater safety and comfort — students don’t have to craft comments that will hold up to public scrutiny, and so typos, errors, and questions are okay!
- There are four types of annotations on Hypothesis: highlights (these are private within a group), annotations (highlighting text + commenting), page notes (commenting on the page without highlighting), and replies to annotations and page notes.
- The Hypothesis sidebar (shown below) can be minimized (click the arrow on the top left of the sidebar) or resized (click the arrow and drag to resize). In the sidebar, you can see the group you are viewing in the middle of the top bar (in this case, HS7310_2019), and clicking the arrow to the right of the group name changes your view (other options include public view or other private groups you belong to). The sidebar shows your own comments and highlights (both private and those shared with the group) and all annotations and page notes your students have shared with the group.
Getting Started: For Educators
- Register for an account.
- Note: The Chrome Plugin allows you to begin annotating any website with one click from your browser bar. The bookmark also allows you to annotate any site, but requires an extra step. You can also add “via.hypothes.is/” to the beginning of a link for students to bring up the annotation tool (e.g. https://via.hypothes.is/pedagogyplayground.com)
- Create a Private Group for your class.
- Copy the Group Link to share with your students.
- Getting Started Guide for Teachers
Setting Students Up For Success
- Students should register for Hypothesis.
- Share the Group Link with students so they can join the group.
- Here’s one example of the type of instructions you might give your students, and here’s another .
- Tips on Annotating in Groups
- Remind students to be attentive to logging into the hypothesis tool and making sure the class group is enabled. Otherwise, it’s easy to accidentally post comments in the public side of hypothesis where the group will not see it. Within the group, it’s also important to post to the whole group vs. leaving private comments. Highlights and private comments within the group can only be seen by the individual who made them.
Assessment:
So how do you assess comments on Hypothesis? I typically assess based on completion. My graduate students, for example, typically read about 7-10 items per week in a Digital Public History class . I ask them to leave a minimum of about 75 words of annotations per reading, and this could take the form of one long comment or two shorter comments. Each week these assignments make up about 2% of each student’s total grade, eventually totaling more than 20% of their final grade. I do not read every comment, but I do read several by each student each week so that I can provide feedback as needed. I also enjoy reading the comments while I read and prepare for class.
Here’s the workflow I use to assess students: I log into hypothesis, click groups in the upper right corner, and select the class I’m assessing.
This takes you to a group page for your class, which looks something like this (see also the screenshot below). On this page, you can see the aggregation of posts your students made WITHIN THE GROUP. (It does not show you their highlights without annotations, private posts to themselves, or comments they accidentally made OUTSIDE the group. ) This page is a great way to quickly gauge class thoughts before or during class since they are sorted by reading (left column in screenshot below) You can also click on each student’s name on the right column (“Members”) to see just that one student’s comments. By selecting each student individually, I can count their comments, briefly skim them, and pick a few to read in depth.
Other Applications:
- Integrate Hypothesis into your LMS : You can integrate Hypothesis into your Learning Management System (e.g. Canvas, Blackboard, etc.). This is a great option if your class reads a lot of PDFs via your LMS.
- 10 Ways to Annotate with Students : This suggests types of annotation that students can do, including doing close reading, doing multimedia analysis by integrating images and videos into annotations, asking questions about the reading, or answering questions pre-composed by the instructor.
- Jeremy Dean and Katherine Schulten, “Skills and Strategies | Annotating to Engage, Analyze, Connect and Create,” The New York Times : Like the link above, this shares additional ways to frame annotation assignments, like annotation as argument, annotating current events to add context and enrich background knowledge, and annotating to make connections.
Resources:
- YouTube Tutorials for Hypothesis
- Teacher Resource Guide : Hypothesis’s curated links for teachers including student examples, assignment examples, and blog posts & webinars.
- Hosting & Annotating PDFs and Really, You Can Annotate Anything : : Tips on turning books, journal articles, and other text into text that can be highlighted and annotated.
- The Pedagogy of Collaborative Annotation : a webinar that includes topics like encouraging student collaboration, scaffolding annotations, annotation assignments and rubrics, and more.
- Meegan Kennedy, “ Open Annotation and Close Reading the Victorian Text: Using Hypothes.is with Students ” (2016): Reflects on the positive benefits of using open annotation in the classroom, but also notes the importance of “careful framing and support.”
If you give Hypothesis Annotations a try, please stop by and comment or email me how it goes for you!
3 thoughts on “ Annotating Readings with Hypothesis ”
This is terrific! @hypothes_is makes it into the #curateteaching keywords under Annotation by Paul Schacht along with a bunch of other actual assignments on annotating a variety of media forms bit.ly/2DTx4nb
I’m blogging for @eCampusSJSU next week on annotation and will be sure to link to this blog post – thanks for putting these up! (This week’s post on getting started in Digital pedagogy: http://blogs.sjsu.edu/ecampus/2019/02/18/but-how-do-i-begin-in-digital-pedagogy/ )
Thanks for sharing such an informational blog which wil, surely be a big help to the students who are creating a thesis or working on dissertation work
- Pingback: how do you annotate a hypothesis - dataslist
Leave a Reply Cancel reply
Your email address will not be published. Required fields are marked *
Notify me of followup comments via e-mail

Hypothes.is Social Annotation
Introduction.
Hypothes.is is an open source tool/plugin for social, collaborative asynchronous annotation . Collaborative annotation is an effective methodology that increases student participation and engagement, expands reading comprehension, and builds critical-thinking skills and community in class. Annotation can also increase faculty, student and cognitive presence in classes. Annotating together makes reading visible, active, and social, enabling students to engage with their texts, teachers, ideas, and each other in deeper, more meaningful ways.
To join our UMB Hypothesis course in Blackboard, where you can experience Hypothesis in a sandbox environment and learn about it, log into Blackboard.umaryland.edu and then come back here and click on this link (won't work if you aren't logged in!).
Basically, Hypothes.is is a layer of conversations on top of the open web and PDF documents. With it, you and your class can:
have private group dialog about open web content (web pages, newspapers, blogs, wikis, etc.)
highlight and take notes on class readings, PDFs and open web content
reply and respond to other group members' comments - have dialog and discussion about readings
check for understanding of concepts in readings
help ensure that students have read and processed readings prior to class
and much more...
How to Get Started - Exploring (Non-Blackboard Integration)
The main thing you need to do to get started is add the extension to your browser. We highly recommend starting with Google Chrome browser, and installing the extension/plug-in from this website . However if you'd prefer to use a different browser, you can also add the Bookmarklet on this page to your browser.
Once you have installed and enabled the Hypothesis extension or bookmarklet, you can click to view the examples below, and see all the annotations on these web pages.
To view the annotation layer on the following examples, you must have the Hypothes.is extension installed and enabled on your browser (see above). Here is an example of a web article with Hypothes.is annotations , and another, a blog post that has annotations , Amanda Gorman's inaugural poem on CNN, or an article on Wired Magazine's website with annotations and discussion. You never know where you might see public annotations on web pages! But all annotations need not be public, you can also annotate privately for yourself, or privately with a group of your choosing.
Blackboard Integration
It is possible to use the Hypothes.is LTI plugin in Blackboard to automatically set up accounts for all instructors and students, as well as a private group for each class that uses it. When Hypothesis is added to a course the app will automatically create a private group for that course. The app will also provision accounts for all students and instructors in the course when they launch the app for the first time. This means that students can begin annotating readings in Blackboard without creating or logging into a separate Hypothesis account. See a video webinar on using Hypothes.is in Blackboard .
These Blackboard-based Hypothesis accounts operate differently than public Hypothesis accounts in the following ways:
They are domain-specific: They only work within the Blackboard instance in which they were created, not on the open web.
They are separate from regular Hypothesis accounts: Users who have created accounts through the public Hypothesis service will be given new and separate accounts for use within the context of Blackboard.
They cannot create their own private groups: A private group is automatically generated for courses where the Hypothesis Blackboard app is installed. Neither instructors nor students will be able to create additional private groups within the Blackboard context.
They do not have access to Hypothesis profile and group activity pages: This is something we plan to address in the near future so that teachers and students can view their annotated comments and conversations outside of the context of a particular document.
Even though these guides are listed for Blackboard they display a Post to Public option when using the tool but since this is installed in Blackboard the only options are to Post to the Course or a Private Post to the user.
Creating Hypothesis-enabled readings in Blackboard
https://web.hypothes.is/help/creating-hypothesis-enabled-readings-in-blackboard/
For a PDF-Based Activity
There are 2 main options for using a PDF for your reading: Upload or use one already in Google Drive, or use one already on the open internet by URL. (This could be a blog post, wiki page, magazine article, etc. ( but with no paywall or login credentials required to view it !) NOTE: It is also possible to use files that are uploaded into Blackboard but it is a bit more convoluted . See the bottom of this page for more info and steps .)
Make sure your PDFs are accessible! This quick OCR tool converts photocopied/scans to accessible text!
For most folks setting up and using a Google Drive account will be the easiest way. Anyone can set up a free account at https://drive.google.com and you can upload your PDFs to a folder that account to get started. We suggest a folder for each class you teach that uses Hypothesis annotation.
Next we will create the Hypothesis Activity item in your course content/modules folder in your Blackboard course:
In a content or module folder, use the Build Content menu to select Hypothesis. Provide a name for your reading/annotation activity, and choose whether or not this is a graded activity (and how many points it is worth, if so.) These are the only 2 settings you need to fill in. Do not upload an attachment here(!) , click Submit to save this item.
Click on the Hypothesis activity you just created in your Bb course and you will see boxes to either Enter URL of web page or PDF , Select PDF from Blackboard or Select PDF from Google Drive . Note: If you choose Select PDF from Blackboard you will need need to take additional steps. Please see the section titled Using Existing Blackboard Course Files below for specific instructions. If you choose Select PDF from Google Drive, please review this article for specific steps.

After you have entered that URL, you are ready to click the item in your course content to launch Hypothesis. All students will have to do is add the Hypothesis extension and click that link to launch the PDF. Below is an image that depicts the things students can do to view annotations, add their own, reply to others, and have dialog about the content:

Grading Student Annotations in Blackboard
https://web.hypothes.is/help/grading-student-annotations-in-blackboard/
Helpful guides to getting started using Hypothesis in Blackboard
https://web.hypothes.is/help/lms-faq/#getting-started
Introduction to the Hypothesis LMS App for Students
https://web.hypothes.is/help/introduction-to-the-hypothesis-lms-app-for-students/
Annotation Tips for Students
https://web.hypothes.is/annotation-tips-for-students/
An Illustrated Taxonomy of Annotation Types
https://web.hypothes.is/blog/varieties-of-hypothesis-annotations-and-their-uses/
Adding Links, Images, and Videos
https://web.hypothes.is/adding-links-images-and-videos/
FAQ for all LMS
https://web.hypothes.is/help/lms-faq/
NOTES on Blackboard Hypothesis Copying from Semester to Semester
Blackboard Hypothesis assignment items (in Blackboard) are separate from the annotation data itself. The former "live" on Bb servers and the latter live on Hypothes.is servers. So, for example, if you delete a Bb Hypothesis reading assignment in Blackboard, that does NOT mean you have deleted the highlights, annotations and discussion. That all lives on Hypothesis servers, separately.
This has a few ramifications:
When you copy a reading assignment from course to course - it sets up a new Hypothesis private group for each course - meaning, it starts fresh, with no annotations or highlights.
If you want to pre-populate a Hypothesis reading with highlights and annotations that you share with all students in your class, you can do so, but when you copy your course to a new semester/section, those annotations will not copy with the Blackboard Hypothesis activity item. There is a separate (and complicated) process for copying Hypothesis annotations and highlights from one section to another (and we don't recommend it.)
We DO recommend providing detailed instructions to students for the Hypothesis activity, that may include screenshots or pointing to specifics in the document - but to do this in the assignment description in Blackboard, rather than using your own highlights/annotations.
Keep in mind, in terms of moving or copying items in Blackboard: if you move a Hypothesis reading activity to another course, the Bb item will copy, but again, NOT the associated annotations.
Non-Blackboard Use
You can also set up a free public Hypothes.is account (not necessary if you use the Blackboard integration above). NOTE: Anyone can create a free Hypothes.is account, but note that if you intend to use the Blackboard Hypothes.is LTI plugin with your UMB classes (info above) that account will be a separate account. So for exploration, we recommend you set up a standard (non-Blackboard) Hypothes.is account with a non-UMB email address (use your gmail, yahoo, hotmail, etc.)
Further Resources
Podcast on social annotation to engage students -(SUNY Oswego-Margaret Schmuhl)
What is Hypothesis, how to use it, and examples -instructor resource page (Carleton College - Wiebke Kuhn)
College resource page for using Hypothesis - instructor resource page (Randolph Macon College- Lily Zhang)
Class Roster & the Social Annotation of Our Names - instructor assignment example (SUNY Finger Lakes CC - Curt Nehring Bliss)
Collaborative Online Annotation with Hypothesis - introduction to Hypothesis module example (Missouri University of Science and Technology - M. Emilia Barbosa and Rachel Schneider) Teaching students to annotate - assignment teaching students to engage in collaborative annotation (Washburn University- Becky Dodge)
The Delftia Project - how to with Hypothesis (North Carolina State University)
Printing readings with annotations - (University of Chicago- Cecilia Lo)
Student Guide to using Hypothesis - (University of California Santa Cruz- Dana Conard)
Active Learning with Collaborative Annotation (CSU Pueblo- Denise Henry)
Promoting Hypothesis after webinar- email to faculty (Middlebury- Bob Cole)
How to OCR a PDF - video (St. Stephen’s School- Jenny Huth)
Using Existing Blackboard Course Files
It is possible to link to existing Blackboard Course files, however there are several steps that are required in order to make this work by creating a course content folder specifically for use with Hypothesis.
In the Blackboard sidebar, under Course Management locate and expand Content Collection and click on your course name.

2. In the Course Content window, click the Create Folder button and give the folder an appropriate name, such as Hypothesis Files and click Submit to create the folder.

3. Set permissions on the folder to allow students to view PDFs. This is done by selecting the small down-arrow to the right of the folder and selecting Permissions from the drop-down menu.

4. Once there, you will click Select Specific Users By Place and choose Course from the dropdown menu.

5. Under Select Roles check the box for Student, and make sure that Read is checked under Set Permissions . Then click the Submit button to save.

Once this is complete, you can upload files to this folder as you would normally. With permissions set the way they are, students will now be able to see these documents when selected for Hypothesis.
Understanding The Difference Between Annotation, Highlights and Page Notes
There are three ways in which you can mark up a document using Hypothesis: Annotations, Highlights or Page Notes. It is important to note that not everyone sees the work that is done using these three different styles.
Annotations
In it's simplest form, annotations are visible to everyone. If you annotate a fragment of a document, the annotation will appear on the right-hand side and will be visible. it is important to note that you can change the privacy of your annotations at any time by clicking on the pencil icon at the bottom right of the annotation card.
A highlight is the digital equivalent of swiping a yellow marker over a passage of text. After you select text on a page, you can use the “Highlight” button in the adder that pops up or press the “h” key on your keyboard. These are always private to you.
A page note is a whole page annotation as opposed to annotating a specific passage of text. These can be made visible or private as you see fit. The intent of a page note is to is to add text and tags as you would an annotation, but on a more global level compared to the granular aspect of a annotation.

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
Margin Size
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
2.4: Hypothesis Annotation
- Last updated
- Save as PDF
- Page ID 75454
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
The following instructions on how to use the Hypothes.is Annotation system are taken directly from the Course Information of a Chemistry class taught by Bob Belford at the University of Arkansas Little Rock.
_____________________________________________
Hypothes.is Web Annotations
This class will use the Hypothes.is web annotation service that is integrated into LibreText. You need to go to https://web.hypothes.is/ and create an account. Please start your username with your last name so you can easily be identified for grading purposes. You should follow it with an underscore and then anything you want, so my username could be belford_123. You will then need to join the class group, which is a private group. You will be sent an email invitation or contact your instructor, and you should only make annotations in the group.
The following YouTube gives a quick demonstration on the use of the Hypothes.is Web Annotation service.
Making Annotations
To make an annotation you simply highlight the text you want to annotate, choose "Annotate" and write in the overlay. Note, the first time you open up a page it will post to "Public" and you need to change that to your class group. Once you have started posting to the class group that will be the default option, but initially the default is public.

If you tag your annotation you can easily find it by going to your homepage in hypothes.is and then filtering by tags. So you may want to make tags like "exam 1", or "nomenclature". If you then filter with your hypothes.is user name and exam 1, you see those items you tagged. If you just do exam 1, you filter for everything the class tagged exam 1. You can then click the contextual link by the annotation, and hypothes.is will open that page, navigate to the highlighted text and display the annotation. (Note: In LibreText the highlights are hidden by default, and you need to click the "eye" with the slash over it (figure \(\PageIndex{2}\)
Using the Hypothes.is Overlay to read discussions
By default the hypothes.is overlay is closed and you do not see the annotations.

Figure \(\PageIndex{2}\) shows how to pick a group and view group activity.

Tags are a very powerful way of organizing your notes and Hypothes.is has different types of tags. Your class group is a tag, and when you click on your hypothes.is group activity (figure \(\PageIndex{2}\)) you only see items posted to your class (note the first tag in the search filter of figure \(\PageIndex{4}\) is a group tag. If you then click your name you see those you have posted to that group. If you remove the group you see all the annotations you have posted anywhere. If you generate a tag based on a topic or exam, you can then easily find that information. Figure \(\PageIndex{4}\) shows a filter using two tags that resulting in four items in a student's "To Do list" for chapter 3 of a cheminformatics class

If the student clicks the contextual link hypothes.is will open the webpage that was highlighted, scroll down to the highlighted section and show the annotation in the overlay. This allows students to organize their notes on LibreText and quickly find content. If you get the chrome plugin you can even annotate material outside of LibreText and connect it to your material and notes within LibreText, as long as the material does not require a login to access. It should also be noted that you can not highlight hidden text within LibreText (like the answers to exercises), and if you have a question on the answer to an exercise, you should highlight the question, not the answer.
Loading metrics
Open Access
Peer-reviewed
Research Article
Leveraging conformal prediction to annotate enzyme function space with limited false positives
Roles Data curation, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing
Affiliation School of Computational Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia, United States of America
Roles Investigation, Methodology, Software, Writing – original draft, Writing – review & editing
Roles Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing
* E-mail: [email protected]
- Kerr Ding,
- Jiaqi Luo,
- Published: May 29, 2024
- https://doi.org/10.1371/journal.pcbi.1012135
- Reader Comments
This is an uncorrected proof.
Machine learning (ML) is increasingly being used to guide biological discovery in biomedicine such as prioritizing promising small molecules in drug discovery. In those applications, ML models are used to predict the properties of biological systems, and researchers use these predictions to prioritize candidates as new biological hypotheses for downstream experimental validations. However, when applied to unseen situations, these models can be overconfident and produce a large number of false positives. One solution to address this issue is to quantify the model’s prediction uncertainty and provide a set of hypotheses with a controlled false discovery rate (FDR) pre-specified by researchers. We propose CPEC, an ML framework for FDR-controlled biological discovery. We demonstrate its effectiveness using enzyme function annotation as a case study, simulating the discovery process of identifying the functions of less-characterized enzymes. CPEC integrates a deep learning model with a statistical tool known as conformal prediction, providing accurate and FDR-controlled function predictions for a given protein enzyme. Conformal prediction provides rigorous statistical guarantees to the predictive model and ensures that the expected FDR will not exceed a user-specified level with high probability. Evaluation experiments show that CPEC achieves reliable FDR control, better or comparable prediction performance at a lower FDR than existing methods, and accurate predictions for enzymes under-represented in the training data. We expect CPEC to be a useful tool for biological discovery applications where a high yield rate in validation experiments is desired but the experimental budget is limited.
Author summary
Machine learning (ML) models are increasingly being applied as predictors to generate biological hypotheses and guide biological discovery. However, when applied to unseen situations, ML models can be overconfident and make enormous false positive predictions, resulting in the challenges for researchers to trade-off between high yield rates and limited budgets. One solution is to quantify the model’s prediction uncertainty and generate predictions at a controlled false discovery rate (FDR) pre-specified by researchers. Here, we introduce CPEC, an ML framework designed for FDR-controlled biological discovery. Using enzyme function prediction as a case study, we simulate the process of function discovery for less-characterized enzymes. Leveraging a statistical framework, conformal prediction, CPEC provides rigorous statistical guarantees that the FDR of the model predictions will not surpass a user-specified level with high probability. Our results suggested that CPEC achieved reliable FDR control for enzymes under-represented in the training data. In the broader context of biological discovery applications, CPEC can be applied to generate high-confidence hypotheses and guide researchers to allocate experimental resources to the validation of hypotheses that are more likely to succeed.
Citation: Ding K, Luo J, Luo Y (2024) Leveraging conformal prediction to annotate enzyme function space with limited false positives. PLoS Comput Biol 20(5): e1012135. https://doi.org/10.1371/journal.pcbi.1012135
Editor: Cameron Mura, University of Virginia, UNITED STATES
Received: September 2, 2023; Accepted: May 3, 2024; Published: May 29, 2024
Copyright: © 2024 Ding et al. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability: The dataset underlying this article was derived from sources in the public domain. We used the data downloaded from https://github.com/flatironinstitute/DeepFRI . Our code is publicly available at https://github.com/luo-group/CPEC .
Funding: This work is supported in part by the National Institute Of General Medical Sciences of the National Institutes of Health ( https://www.nih.gov/ ) under the award R35GM150890, the 2022 Amazon Research Award ( https://www.amazon.science/research-awards ), and the Seed Grant Program from the NSF AI Institute: Molecule Maker Lab Institute (grant #2019897) at the University of Illinois Urbana-Champaign (UIUC; https://moleculemaker.org/ ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have declared that no competing interests exist.
Introduction
Machine learning (ML) algorithms have proven to be transformative tools for generating biological hypotheses and uncovering knowledge from large datasets [ 1 , 2 ]. Applications include designing function-enhanced proteins [ 3 , 4 ], searching for novel drug molecules [ 5 ], and optimizing human antibodies against new viral variants [ 6 ]. These discoveries often involve a combination of computation and experimentation, where ML-based predictive models generate biological hypotheses and wet-lab experiments are then used to validate them. This approach is beneficial as it greatly reduces the search space and eliminates candidates that are unlikely to be successful, thus saving time and resources in the discovery process. For example, in drug discovery, ML has become a popular strategy for virtual screening of molecule libraries, where researchers use ML models to predict the properties of molecules, such as binding affinity to a target, and identify the most promising candidates for downstream experimental validation and lead optimization [ 7 ].
To gain new insights into biological systems or make novel discoveries (e.g., designing new drugs), ML algorithms are often used to make predictions for previously unseen data samples. For example, to support the design of new vaccines or therapeutics for COVID-19, ML algorithms need to predict the potential for immune escape of future variants that are composed of mutations that have not yet been seen. Similarly, in drug screening, ML algorithms should be able to predict molecules that are structurally different from those in the training data, which helps scientists avoid re-discovering existing drugs. However, making predictions for samples that are under-represented in the training data is a challenging task in ML. While human experts can assess the success likelihood of generated hypotheses based on their domain knowledge or intuition, this ability is not naturally developed by an ML model and, as a result, the model could be susceptible to pathological failure and only provide overconfident or unreliable predictions. This can have critical implications in ML-assisted biological discovery, as unreliable ML predictions can guide experimental efforts in the wrong direction, wasting resources on validating false positives.
In this work, we aim to develop ML models that can generate hypotheses with limited false positives, providing confident and accurate predictions that can potentially help improve the yield rate in downstream validation experiments. Specifically, we use the function annotation problem of protein enzymes as an example to demonstrate our method. The underlying computational problem of function annotation is a multi-class, multi-label classification problem as a protein can have multiple functions. In computational protein function annotation, a model typically predicts a set of functions that the query protein may potentially have. The set of predicted functions, if validated by experiments, can be incorporated into existing databases to augment our knowledge of the protein function space. There is often a trade-off regarding the size of the prediction set: researchers prefer a set with a small size, containing a handful of very confident predictions, as it is not desirable to spend resources on too many hypotheses that ultimately turn out to be false positives; on the other hand, researchers may be willing to increase the budget to validate a larger set of predictions in order to improve the chance of discovering novel functions for under-studied proteins.
The above tradeoff is often captured by different notions of prediction score cutoff, which decides whether to assign a particular function label to a protein, in existing computational methods for function annotation. For example, when annotating protein functions using sequence-similarity-based tools such as BLAST [ 8 ], a cutoff of the BLAST E-value can be used to determine the significance of sequence match. However, the choice of E-value cutoff is often based on the user’s intuition and good cutoff values on a dataset may not generalize to another dataset. Recent ML methods for enzyme function annotation typically first predict the probability that the input protein has a particular function and annotate the protein with this function if the predicted probability is greater than 0.5 [ 9 – 11 ]. However, using an arbitrary cutoff such as 0.5 is problematic as the predicted probabilities do not always translate to the confidence of the ML model, especially when the model is not well-calibrated (e.g., a predicted function with probability 0.95 may still be an unreliable prediction if the model is overconfident and produces very high probability scores most of the time). Recently, Hie et al. [ 12 ] developed a framework that used the Gaussian process to estimate the confidence or uncertainty in the ML model’s predictions. While the framework was shown to be effective to guide biological discovery, it is unclear how the estimated uncertainty is related to the final false discovery rate (FDR) in experimental validation and how to set a cutoff on the uncertainty scores to achieve a desired FDR. Consequently, it is challenging to provide FDR estimates before the experimental validation, and FDR typically can only be assessed post-validation.
Here, we propose an ML method, called CPEC (Conformal Prediction of EC number), to achieve FDR-controlled enzyme function prediction by leveraging a statistical framework known as conformal prediction (CP) [ 13 ]. CPEC receives the sequence or structure of an enzyme as input and predicts a set of functions (EC numbers) that the enzyme potentially has. The unique strength of CPEC is that the averaged per-protein FDR (i.e., the number of incorrect predictions divided by the prediction set size for a protein) can be controlled by a user-specified hyper-parameter α . The CP framework theoretically guarantees that the FDR of our per-protein predictions is no larger than α with a very high probability. This equips researchers with foresight, offering success rate estimates even before experimental validation. In an ML-guided workflow of protein function discovery, researchers can specify the desired FDR level α based on the experiment budget or expectations. For example, setting α to a smaller value when only the most confident predictions are needed or the test budget is limited, or setting to a larger value when the goal is to discover novel functions and a slightly higher FDR and budget are acceptable. The base ML model of CPEC is PenLight2, an improved version of the deep learning model PenLight [ 14 ] for the multi-class multi-label protein function annotation problem, which uses a graph neural network to integrate 3D protein structure data and protein language model embeddings to learn structure-aware representations for function prediction. Benchmarked on a carefully curated dataset, we first found that CPEC outperformed existing deep learning methods for enzyme function prediction. We also demonstrated that CPEC provides rigorous guarantees of FDR and allows users to trade-off between precision and recall in the predictions by tuning the desired maximum value α of FDR. Additionally, we showed that CPEC consistently provides FDR-controlled predictions for proteins with different sequence identities to the training set, suggesting its robustness even in regimes beyond its training data distribution. Moreover, based on CPEC, we proposed a cascade model that can better balance the resolution and coverage for EC number prediction.
Materials and methods
Problem formulation.
- PPT PowerPoint slide
- PNG larger image
- TIFF original image
(A) CPEC is a machine learning (ML) framework that leverages conformal prediction to control the false discovery rate (FDR) while performing enzyme function predictions. Compared to conventional ML predictions, CPEC allows users to select the desired FDR tolerance α and generates corresponding FDR-controlled prediction sets. Enabled by conformal prediction, CPEC provides a rigorous statistical guarantee such that the FDR of its predictions will not exceed the FDR tolerance α set by the users. The FDR tolerance α offers flexibilities in ML-guided biological discovery: when α is small, CPEC only produces hypotheses for which it has the most confidence; a larger α value would allow CPEC to afford a higher FDR, and CPEC thus can predict a set with more function labels to improve the true positive rate. Abbreviation: Func: function. Incorrect predictions in prediction sets are colored gray. (B) We developed a deep learning model, PenLight2, as the base model of the CPEC framework. The model is a graph neural network that receives the three-dimensional structure and the sequence of a protein as input and generates a function-aware representation for the protein. It employs a contrastive learning scheme to learn a vector representation for proteins, such that the representations of functionally similar proteins in the latent space are pulled together while dissimilar proteins are pushed apart.
https://doi.org/10.1371/journal.pcbi.1012135.g001
Conformal risk control
Overview of conformal risk control..
Conformal risk control guarantee for FDR control.
Calibration algorithm for FDR control.
Given the FDR control guarantee, the natural follow-up question would be how to find a valid parameter λ that can control the risk through the calibration step on calibration data. The Learn then Test (LTT) algorithm [ 22 ], which formulated the selection of λ as a multiple hypotheses testing problem, has been proposed to solve this question. CPEC adopts the LTT algorithm established upon the data distribution assumption that all feature-response pairs ( X , Y ) from the calibration set and the test set are independent and identically distributed (i.i.d.).
Algorithm 1: CPEC for FDR control
/* Calculation of Hoeffding’s inequality p-values { p 1 , …, p N } */
for i ← 1 N do
for j ← 1 n c do
while p i ≤ δ and i ≥ 1 do
i ← i − 1;
Protein function prediction
Ec number prediction dataset..
We applied CPEC on the task of Enzyme Commission (EC) numbers [ 17 ] prediction to demonstrate its effectiveness. EC number is a widely used four-level classification scheme, which organizes the protein according to their functions of catalyzing biochemical reactions. In specific, a protein would be labeled with an EC number if it catalyzes the type of biochemical reactions represented by that EC number. For each four-digit EC number a . b . c . d , the 1st-level a is the most general classification level while the 4th-level d is the most specific one. We used the dataset that contains EC number-labeled protein sequences and structures, provided by Gligorijević et al. [ 10 ]. The protein structures were retrieved from Protein Data Bank (PDB) [ 27 ]. Protein chains were then clustered at 95% sequence identity using the BLASTClust function in the BLAST tool [ 8 ] and then organized into a non-redundant set which only included one labeled high-resolution protein chain from each cluster. The EC number annotations were collected from SIFTS (structure integration with function, taxonomy, and sequence) [ 28 ]. As the 4th-level EC number is the most informative functional label, we only kept proteins that have ground-truth level-4 EC numbers in our experiments. Eventually, the dataset we used has 10, 245 proteins and a train/valid/test ratio of roughly 7: 1: 2. The proteins in the test set have a maximum sequence identity of 95% to the training set. Within the test set, test proteins were further divided into disjoint groups with [0, 30%), [30%, 40%), [40%, 50%), [50%, 70%), and [70%, 95%] sequence identity to the training set. The lower the sequence identity to the training set, the more difficult the test protein would be for ML models to predict its functions. In experiments, we have used the more challenging test data group ([0, 30%)) to evaluate the robustness of our framework.
Contrastive learning-based protein function prediction.
For protein function prediction tasks, supervised learning has long been a popular choice in the deep learning community. Supervised learning-based methods take protein sequences or structures as input and directly map them into class labels. While the idea is simple and efficient, supervised learning has been suffering from a major drawback: its performance could be severely affected by the class imbalances of the training data, an unfortunately common phenomenon in protein function prediction tasks. For example, in the EC number database, some EC classes contain very few proteins (less than ten), while some other EC classes contain more than a hundred proteins. Those classes with more proteins would dominate the training, thereby suppressing the minority classes and degrading the performance of supervised learning. To overcome this challenge, a new paradigm called contrastive learning has become popular in recent years [ 29 ]. Instead of directly outputting class labels, contrastive learning-based models map the training proteins into an embedding space where functionally similar proteins are close to each other and functionally dissimilar pairs are far away. Our previously developed ML methods PenLight and CLEAN [ 14 , 30 ] have demonstrated the effectiveness of contrastive learning in enzyme function predictions. In each iteration of the contrastive learning process, the PenLight or CLEAN model samples a triplet including an anchor protein p 0 , a positive protein p + , and a negative protein p − , such the positive protein pairs ( p 0 , p + ) have similar EC numbers (e.g., under the same subtree in the EC number ontology) while the negative pairs ( p 0 , p − ) have dissimilar EC numbers. The objective of contrastive learning is to learn low-dimensional embeddings x 0 , x + , x − for the protein triplet such that the embedding distance d ( x 0 , x + ) is minimized while d ( x 0 , x − ) is maximized ( Fig 1B and S1 Text ). In the prediction time, the EC number of the training protein with the closest embedding distance to the query protein will be used as the predicted function labels for the query protein.
In this work, we developed PenLight2, an extension of our previous PenLight model [ 14 ] for performing multi-label classification of EC numbers. Similar to PenLight, PenLight2 is a structure-based contrastive learning framework that integrates protein sequence and structure data for predicting protein function. It integrated protein 3D structures and protein language model (ESM-1b [ 31 ]) embeddings into a graph attention network [ 32 ] and optimized the model using the contrastive learning approach, which pulled the embeddings of the (anchor, positive) pair together and the embeddings of the (anchor, negative) pair away. By naturally representing the amino acids as nodes and spatial relations between residues as edges, the graph neural network can extract structural features in addition to sequence features and generate function-aware representations of the protein. In this work, we shifted from the multi-class single-label classification approach used in PenLight [ 14 ] to a multi-class multi-label classification framework, which better aligns with the function annotation data of enzymes in which an enzyme can be labeled with multiple EC numbers. PenLight2 achieved two key improvements compared to PenLight: model training (triplet sampling strategy) and model inference (function transfer scheme and prediction cutoff selection):
1) Triplet sampling strategy. For training efficiency, PenLight takes a multi-class single-label classification approach and randomly samples one EC number for promiscuous enzymes when constructing the triplet in contrastive learning, considering that only less than 10% enzymes in the database used are annotated with more than one EC number. To enhance the effectiveness of contrastive learning for promiscuous enzymes, in this work, we adopt a multi-class multi-label classification approach, in which retain the complete four-level EC number annotations for an enzyme in the triplet sampling of PenLight2 ( Fig 1B ). Specifically, we thus generalized PenLight’s hierarchical sampling scheme to accommodate proteins with multiple functions in PenLight2: in each training epoch, for each anchor protein (every protein in the training set), we randomly choose one of its ground truth EC numbers if it has more than one and then follow original sampling scheme in PenLight for the sampling of the positive and the negative proteins ( S1 Text ). A filter is applied to ensure that the anchor and the negative do not share EC numbers.
3) Prediction cutoff selection. In contrast to the original PenLight model that only predicted the top-1 EC number for a query protein, PenLight2 implemented an adaptive method to achieve multi-label EC prediction. Following the max-separation method proposed in our previous study [ 30 ], we sorted the distances between the query protein and all EC clusters and identified the max difference between adjacent distances. PenLight2 then uses the position with the max separation as the cutoff point and outputs all EC numbers before this point as final predictions. This cutoff selection method aligns with the multi-label nature of the task.
With these improvements, we extended the original PenLight from the single-label classification to the multi-label setting. We denote this improved version as PenLight2.
We performed multiple experiments to evaluate CPEC’s prediction accuracy and ability of FDR control. We further evaluated CPEC using test data that have low sequence identities to the training data to demonstrate its utility for generating hypotheses (function annotations) for novel protein sequences.
CPEC achieves accurate enzyme function predictions
We first evaluated the prediction performance of PenLight2, the base ML model in CPEC, for predicting function annotations (EC numbers) of protein enzymes. The purpose of this experiment was to assess the baseline prediction accuracy of CPEC when the FDR control is not applied. We compared CPEC with three state-of-the-art deep learning methods capable of reliably predicting enzyme function on the fourth level of EC hierarchy, including two CNN-based (convolutional neural networks) methods DeepEC [ 9 ] and ProteInfer [ 11 ] that take protein sequence data as input and one GNN-based (graph neural networks) method DeepFRI [ 10 ] that takes both protein sequence and structure data as input. All these three methods applied the multi-class classification paradigm for function prediction: first predicting a score between 0 and 1 as the predicted probability that the input enzyme has a particular EC number and then generating all EC numbers with predicted probability greater than 0.5 (except for DeepFRI which used 0.1 as cutoff) as the final predicted function annotations for the input enzyme. We evaluated all methods using metrics F1 score, which assesses prediction accuracy considering both precision and recall, and the normalized discounted cumulative gain (nDCG) [ 33 ], which rewards higher rankings of true positives over false negatives in the prediction set ( S1 Text ). On a more challenging test set (test proteins with [0, 30%) sequence identity to training proteins), we further evaluated all methods by drawing the micro-averaged precision-recall curves.
The evaluation results showed that our method outperformed all the three state-of-the-art methods in terms of both F1 score and nDCG ( Fig 2A ). For example, PenLight2 achieved a significant improvement of 34% and 26% for F1 and nDCG, respectively, over the second-best baseline DeepFRI. The pronounced performance gaps between PenLight2 and other baselines also suggested the effectiveness of the contrastive learning strategy used in PenLight2. The major reason is that contrastive learning utilized the structure of the function space (the hierarchical tree of the EC number classification system) to learn protein embeddings that reflect function similarity, while the multi-class classification strategy used in the three baselines just treated all EC numbers as a flat list of labels and may only capture sequence/structure similarity but not function similarity. In addition, we observed that methods that incorporated protein structure data (PenLight2 and DeepFRI) achieved better than methods that only use sequence data as input (DeepEC and ProteInfer), suggesting that protein structure may describe features related to functions more explicitly and is useful for predicting protein function. Those results demonstrated that the design choices of PenLight2, including the contrastive learning strategy and representation learning of protein structure, greatly improve the accuracy of protein function prediction. To further analyze PenLight2’s prediction performance, we delineated its F1 score into precision and recall and observed that PenLight2 has slightly lower precision than other methods but substantially higher recall and F1 score ( S1 Fig ). We noted that other baseline methods such as ProteInfer achieved the high precision score at a cost of low coverage ( Fig 2A ), meaning that they did not predict any functions for a large number of query proteins due to their high uncertainties in those proteins. Additionally, we evaluated PenLight despite that it only performs single-label prediction, and we found that PenLight and PenLight2 had similar performances. As the fraction of promiscuous enzymes is low in the test set, we expected PenLight2 to be a more accurate predictor than PenLight in future enzyme function prediction tasks when promiscuous enzymes prevail.
(A) We evaluated DeepEC [ 9 ], ProteInfer [ 11 ], DeepFRI [ 10 ], and PenLight2 for predicting the 4th-level EC number, using F1 score, the normalized discounted cumulative gain (nDCG), and coverage as the metrics. Specifically, coverage is defined as the proportion of test proteins for which a method has made at least one EC number prediction. (B) We further evaluated all methods for predicting the 4th-level EC number on more challenging test proteins with [0, 30%) sequence identities to the training proteins and drew the micro-averaged precision-recall curves. For each curve, we labeled the point with the maximum F1 score (Fmax).
https://doi.org/10.1371/journal.pcbi.1012135.g002
On a more challenging test set which only includes test proteins with [0, 30%) sequence identities to training proteins, we also observed that PenLight2 robustly predicted the EC numbers of the test proteins and outperformed all baseline methods ( Fig 2B and S3 Fig ). The improvement of the micro-averaged Fmax value from the best baseline method ProteInfer to PenLight2 was 32%. In the high recall region, PenLight2 achieved a higher precision value than any of the baseline methods. The results here were consistent with the results on the entire test set, which further proved the effectiveness of PenLight2 for EC number prediction.
CPEC provides FDR control for EC number prediction
After validating its prediction performance, we integrated PenLight2 as the base model into the conformal prediction framework. Conformal prediction provides a flexible, data-driven way to find an optimal cutoff for PenLigth2 to decide whether to predict a function label for the input protein, such that the FDR on the test data is lower than the user-specified FDR upper bound α . Here, we performed experiments to investigate whether CPEC achieves the desired FDRs and how its prediction performance would change when varying α . For comparison, we compared CPEC to several other thresholding strategies for generating the prediction set, including 1) max-separation ( Methods ); 2) top-1, where only the EC number with the closest embedding distance to the input protein is predicted as output; and 3) σ -threshold, where all EC numbers with an embedding distance smaller than μ + 2 σ to the input protein are predicted as output, where μ and σ are the mean and standard deviation of a positive control set that contains the distances between all true protein-EC number pairs. Platt scaling [ 34 ], a parametric calibration method, was further included as a thresholding strategy for comparison. We also included our baseline DeepFRI, which outputs EC numbers if it predicts that the probability of the input having this EC number is greater than a cutoff of 0.1. The purpose of the experiment here is not to show CPEC can outperform all other methods under all metrics but to show that CPEC can achieve a desired tradeoff by tuning the interpretable parameter α and simultaneously provide a rigorous statistical guarantee on its FDR. In an evaluation experiment, we have further compared CPEC with two point-uncertainty prediction methods (Monte Carlo dropout [ 35 ] and RED [ 36 ]), demonstrating that CPEC provides precise FDR control prior to validation, whereas MC dropout and RED can only evaluate FDR post-validation ( S1 Text ).
Reliable FDR controls.
In theory, the conformal prediction framework guarantees that the actual FDR of the base ML model on the test data is smaller than the pre-specified FDR level α with high probability. We first investigated how well this property holds on our function prediction task. We varied the value of α from 0 to 1, with increments of 0.1, and measured CPEC’s averaged per-protein FDR on the test data. As expected, we observed that the actual FDR of CPEC ( Fig 3A , blue line) was strictly below the specified FDR upper bound α ( Fig 3A , diagonal line) across different α values. This result suggested that CPEC successfully achieved reliable FDRs as guaranteed by the conformal prediction. We have further performed an evaluation experiment to investigate the impact of the calibration set sizes on CPEC’s FDR control, and the results suggested that the FDR control performances of CPEC were robust to various calibration set sizes ( S1 Text ).
For FDR tolerance α from 0.1 to 0.9 with increments 0.1, we evaluated how well CPEC controls the FDR for EC number prediction. Observed FDR risks, precision averaged over samples, recall averaged over samples, F1 score averaged over samples, and nDCG were reported for each FDR tolerance on test proteins in (A-E). The black dotted line in (A) represents the theoretical upper bound of FDR over test proteins. Three thresholding strategies were assessed over PenLight2 as a comparison to CPEC, which includes 1) max-separation [ 30 ], 2) top-1, and 3) σ -threshold. The results of CPEC were averaged over five different seeds. DeepFRI was also included for comparison.
https://doi.org/10.1371/journal.pcbi.1012135.g003
Tradeoff between precision and recall with controlled FDR.
Varying the FDR parameter α allowed us to trade-off between the prediction precision and recall of CPEC ( Fig 3B and 3C ). When α was small, CPEC predicted function labels for which it has the most confidence, in order to achieve a lower FDR, resulting in high precision scores (e.g., precision 0.9 when α = 0.1). When CPEC was allowed to tolerate a relatively larger FDR α , it predicted more potential function labels for the input protein at the FDR level it can afford, which resulted in an increasing recall score as α was increasing. Similarly, the nDCG score of CPEC was also increasing with α ( Fig 3E ), indicating that CPEC not only retrieved more true function labels but also ranked the true labels at the top of its prediction list.
Interpretable cutoff for guiding discovery.
CPEC is able to compute an adaptive cutoff internally based on the user-specified FDR parameter α for deciding whether or not to assign a function label to the input protein. This allows researchers to prioritize or balance precision, recall, and FDR, depending on test budget or experiment expectations, in an ML-guided biological discovery process. In contrast, many existing methods that use a constant cutoff often have optimized performance in one metric but suffer in another. For example, in our experiment, DeepFRI and Platt scaling threshold strategy had the highest precisions but their recalls were the lowest among all methods; the σ -threshold strategy had a recall of 0.94 yet its FDR (0.75) was substantially higher than others ( Fig 3A–3C and S2 Fig ). Although some methods such as DeepFRI may achieve a better tradeoff between precision and recall by varying its probability cutoff from 0.1 to other values, they lack a rigorous statistical guarantee on the effect of varying the cutoff values. For example, if the cutoff of DeepFRI was raised from 0.1 to 0.9, one can expect that, qualitatively, it would lead to a higher precision but also a higher FDR. However, it is hard to quantitatively interpret the consequence of raising the cutoff to 0.9 (e.g., how high would the FDR be) until the model is evaluated using ground-truth labels, which are often unavailable before experimental validation in the process of biological discovery. In contrast, with CPEC, researchers are also able to balance the interplay between the prediction precision and recall by tuning the interpretable parameter α and assured that the resulting FDR will not be greater than α .
Overall, through these experiments, we validated that CPEC can achieve the statistical guarantee of FDR. We further evaluated the effect of varying the FDR tolerance α on CPEC’s prediction performances. Compared to conventional strategies for multi-label protein function prediction, CPEC provides a flexible and statistically rigorous way to better tradeoff precision and recall, which can be used to better guide exploration and exploitation in biological discovery with a controlled FDR.
Adaptive prediction of EC numbers for proteins with different sequence identities to the training set
The risk in our conformal prediction framework is defined as the global average of per-protein FDRs, which may raise the concern that the overall FDR control achieved by CPEC on the test set was mainly contributed by FDR controls on those proteins that are easy to characterize and predict, and it is possible that the model suffered from pathological failures and did not give accurate FDR controls on proteins that are hard to predict. To this point, we defined the prediction difficulty based on the level of sequence identity between test proteins and training proteins, following the intuition that it is more challenging for an ML model to predict the functions of a protein if the protein does not have homologous sequences in the training data. We first performed a stratified evaluation to analyze CPEC’s FDR-control performance at different levels of prediction difficulty. After examining the consistency of the FDR control across different difficulties, we explored an adaptive strategy for predicting EC numbers, which allows the ML model not to predict a too specific EC number than what the evidence supported and only predict at the most confident level of EC hierarchy.
Consistency of FDR control.
We first confirmed CPEC’s FDR-control ability across different levels of prediction difficulty. Specifically, we partitioned the test set into disjoint groups based on the following ranges of sequence identity to the training set: [0, 30%), [30%, 40%), [40%, 50%), [50%, 70%) and [70%, 95%]. We varied the values of α from 0.05 to 0.5 with increments of 0.05. For each level of FDR tolerance α , we examined the FDR within each group of test proteins. As shown in Fig 4A , CPEC achieved consistent FDR controls across different levels of train-test sequence identity and different values of α , where the observed FDR were all below the pre-specified FDR tolerance α . Even for the most difficult group of test proteins that only have [0, 30%) sequence identity to the training proteins, CPEC still achieved an FDR of 0.03 when tolerance α = 0.1. This is because a well-trained ML model would have low confidence when encountering difficult inputs, and CPEC would abstain from making predictions if the model’s confidence does not exceed the decision threshold. The results of this experiment built upon the conclusion of the previous subsection and validated that CPEC can not only control the FDR of the entire test set but also the FDR for each group of test proteins with different levels of prediction difficulty. We have performed an evaluation experiment to further assess CPEC’s FDR control on test proteins that do not belong to the same CATH superfamilies [ 37 ] as any of the training proteins. We found that CPEC provided effective FDR control for these test proteins from unseen superfamilies ( S5 – S7 Figs and S1 Text ), suggesting that CPEC can offer effective FDR-controlled EC number predictions even for test proteins that are very dissimilar to its training proteins.
(A) We reported the observed FDR for test proteins with different sequence identities to the training set (i.e. different difficulty levels) for FDR tolerance α from 0.05 to 0.5 with increments of 0.05. Test proteins were divided into disjoint groups with [0, 30%), [30%, 40%), [40%, 50%), [50%, 70%), and [70%, 95%] sequence identity to the training set. The smaller the sequence identity, the harder the protein would be for machine learning models to predict function labels. (B) We designed the procedure to first predict the EC number at the 4th level. If the model was uncertain at this level and did not make any predictions, we would move to the 3rd level to make more confident conformal predictions instead of continuing with the 4th level with high risks. We used the same FDR tolerance of α = 0.2 for both levels of CPEC prediction. For proteins with different sequence identities to the training data, we reported the hit rate of our proposed procedure. The hit rate on the 4th level, the hit rate on the 3rd level, the percentage of proteins with incorrect predictions on both levels, and the percentage of not called proteins for both levels were reported. The results were calculated as an average over 5 different seeds of splitting the calibration set.
https://doi.org/10.1371/journal.pcbi.1012135.g004
An adaptive strategy for EC number prediction.
The EC number hierarchy assigns four-digit numbers to enzymes, where the 4th-level label describes the most specific functions of enzymes whereas the 1st-level label describes the most general functions. In EC number prediction, ideally, a predictive model should not predict a too specific EC number than what the evidence supported. In other words, if a model is only confident about its prediction up to the 3rd level of an EC number for a protein, it should not output an arbitrary prediction at the 4th level. We first trained two CPEC models, where the first model, denoted as CPEC4, predicts EC numbers at the 4th level as regular, and the other, denoted as CPEC3, predicts the 3rd-level EC numbers. We then combine the two models as a cascade model: given an input protein and a desired value of α , we first apply the CPEC4 to predict the 4th-level EC numbers for the input protein with an FDR at most α . If CPEC4 outputs any 4th-level EC numbers, they will be used as the fine-level annotations for the input; if CPEC4 predicts nothing due to the FDR tolerance α being too stringent, we apply CPEC3 on the same input to predict EC numbers at the 3rd-level. If CPEC3 outputs any 3rd-level EC numbers, they will be used as the coarse-level annotations for the input; otherwise, the cascade model just predicts nothing for this input. The motivation of this adaptive prediction strategy is that even though 3rd-level EC numbers are less informative than 4th-level ones, it might be more useful for researchers in certain circumstances to acquire confident 3rd-level EC numbers than only obtaining a prediction set with a large number of false positive EC numbers at the 4th level.
To validate the feasibility of the above adaptive model, we evaluated CPEC3 and CPEC4 using the same FDR tolerance α = 0.2 on our test set. We reported the hit rate, defined as the fraction proteins for which our model predicted at least one correct EC number, for both the 4th-level and the 3rd-level EC numbers. We found that this adaptive prediction model, compared to the model that only predicts at the 4th level, greatly reduced the number of proteins for which the model made incorrect predictions or did not make predictions ( Fig 4B ). For example, on the test group with sequence identity [0, 30%) to the training data, around 60% proteins were correctly annotated with at least one EC number, while only 40% proteins were correctly annotated if the adaptive strategy was not used. This experiment demonstrated the applicability of CPEC for balancing the prediction resolution and coverage in protein function annotation.
Application: EC number annotation for low-sequence-identity proteins
Conformal prediction quantifies the ML model’s uncertainty in its predictions, especially for the predictions for previously unseen data. This is extremely useful in ML-guided biological discovery as we often need to make predictions for unseen data to gain novel discoveries. For example, in protein function annotation, the most challenging proteins to annotate are those previously uncharacterized or do not have sequence homologous in current databases. Conventional ML models that do not quantify prediction uncertainties are often overconfident when making predictions for the aforementioned challenging samples, leading to a large number of false positives in their predictions, which can incur a high cost in experimental validation without yielding a high true positive rate. Considering the importance of predicting previously uncharacterized data, here we designed an evaluation experiment to assess CPEC’s prediction performance on these challenging proteins. We created a test set that contains only proteins that have less than 30% sequence identity to any proteins in the training set, which simulated a challenging application scenario.
We varied the FDR tolerance α from 0.05 to 0.5 and counted the number of correct predictions, where assigning one EC number to a protein was counted as one prediction. We observed that CPEC had an effective uncertainty quantification for its predictions on this low-sequence-identity test set ( Fig 5A ). For example, when α = 0.05 which forced the model to only output the most confident predictions, CPEC was highly accurate, with a precision of nearly 0.97. At the FDR tolerance level of 0.1, CPEC was able to retrieve 25% (180/777) true protein-EC number pairs at a precision higher than 0.9. Keeping increasing the value of α allowed CPEC to make more correct predictions, without significantly sacrificing precision. For instance, at the level α = 0.5, CPEC successfully predicted 70% true protein-EC number pairs while maintaining a reasonable precision of 0.6 and an nDCG of 0.7. As a comparison, the baseline method DeepFRI correctly predicted 309 protein-EC pairs, out of the total 777 true pairs, with a precision of 0.89 and an nDCG score of 0.50, which roughly corresponds to CPEC’s performance at α = 0.2.
(A) CPEC was evaluated on difficult test proteins ([0, 30%) sequence identity to the training data). For FDR tolerance from 0.05 to 0.5, the total number of correct predictions, precision averaged over samples, and the normalized discounted cumulative gain was reported under five different seeds for splitting calibration data. Note that the upper bound of correct predictions, i.e. the ground truth labels, is 777. As a comparison, DeepFRI successfully made 307 predictions, with a sample-averaged precision of 0.8911 and an nDCG score of 0.5023. (B) An example of the prediction sets generated by CPEC for Gag-Pol polyprotein (UniProt ID: P04584; PDB ID: 3F9K), along with the prediction set from DeepFRI. CPEC used the chain A of the PDB structure as input. The prediction sets were generated under FDR tolerance α = 0.25, 0.3, 0.35. The sequence of this protein has [0, 30%) sequence identity to the training set and, therefore, can be viewed as a challenging sample. Incorrect EC number predictions are colored gray. (C) Boxplots showing the FDR@1st-hit metric, defined as the smallest FDR tolerance α at which CPEC made the first correct prediction for each protein. The evaluation was performed on five groups of test proteins, stratified based on their sequence identities to the training set.
https://doi.org/10.1371/journal.pcbi.1012135.g005
We again note that CPEC is more flexible than methods such as DeepFRI in that it provides an interpretable and principled way to tradeoff between precision and recall, which allows researchers to not only prioritize high-confidence predictions but also increase prediction coverage for improving the yield rate of true positives. To illustrate this, we visualized the prediction results of CPEC and DeepFRI in Fig 5B . We selected a protein that has multiple EC number annotations (UniProt ID: P04584). Using its default setting, DeepFRI predicted four labels for this protein, among which three were correct. For CPEC, we gradually increased the value of α and see how the prediction set was changing. Interestingly, we observed that CPEC gradually predicted more true EC numbers as α was increasing while maintaining a low FDR. In particular, when α = 0.25, CPEC outputted two EC numbers, both of which were correct predictions; when α was relaxed to 0.3, CPEC predicted one more EC number, which turned out to be also correct; when we further relaxed the FDR tolerance α to 0.35, CPEC predicted six EC numbers for the protein, and four of them were correct. This example illustrated CPEC’s utility in practice: researchers have the flexibility when using CPEC to guide experiments, where a small value of α prioritizes accurate and confident hypotheses, and a large value of α promotes a high yield of true positives while ensuring the number of false positives to be limited.
Having observed that CPEC was able to recover more true function labels as we were relaxing the FDR tolerance α , we asked one important question—at which value of α can CPEC output the first correct function label (“hit”) for the input protein. We referred to this α value as FDR@1st-hit. This metric can be viewed as a proxy of the experiment cost researchers need to pay before they obtain the first validated hypothesis. We computed the FDR@1st-hit value for all test proteins in each of the five disjoint groups partitioned by their sequence identity to training sequences ( Fig 5C ). We found that for the majority of the test sequences (the four groups out of five with sequence identity at least 30% to training sequences), CPEC was able to reach the first hit at an FDR lower than 0.15. For the most difficult group where all proteins share [0, 30%) sequence identity to training data, the median FDR@1st-hit was 0.3. This observation was consistent with our intuition and expectation, as low-sequence-identity proteins are more difficult for the ML model to predict, thus requiring a larger hypotheses space to include at least one true positive. Overall, CPEC achieved a reasonable FDR@1st-hit for function annotation, meaning that it produced a limited number of false positives before recovering at least one true positive, which is a highly desired advantage in ML-guided biological discovery.
Machine learning models play a vital role in generating biological hypotheses for downstream experimental analyses and facilitating biological discoveries in various applications. A significant challenge in the process of ML-assisted biological discoveries is the development of ML models with interpretable uncertainty quantification of predictions. When applied to unseen situations, ML models without uncertainty quantification are susceptible to overconfident predictions, which misdirects experimental efforts and resources to the validation of false positive hypotheses. Addressing this challenge becomes essential to ensure the efficiency and reliability of ML-assisted biological discovery.
In this work, we have presented CPEC, an ML framework that enables FDR-controlled ML-assisted biological discoveries. Leveraging the conformal prediction framework, CPEC allows users to specify their desired FDR tolerance α , tailored to the experiment budget or goals and makes corresponding predictions with a controlled FDR. We demonstrate CPEC’s effectiveness using enzyme function annotation as a case study, simulating the discovery process of identifying the functions of less-characterized enzymes. PenLight2, an improved version of PenLight optimized for multi-label classification is utilized as CPEC’s base ML model. Specifically, CPEC takes the sequence and structure of an enzyme as input and outputs a set of functions (EC numbers) that the enzyme potentially has. The conformal prediction algorithm in CPEC theoretically guarantees that the FDR of the predicted set of functions will not exceed α with high probability. The evaluation of CPEC on the EC number prediction task showed that CPEC provides reliable FDR control and has comparable or better prediction accuracy than baseline methods at a much lower FDR. Interpretable cutoffs were provided by CPEC for guiding the EC number annotations of proteins. Furthermore, CPEC demonstrated its robustness in making FDR-controlled predictions even for proteins with low sequence identity to its training set.
Quantifying uncertainties of ML model predictions is a key desideratum in ML-guided biological discovery. Although a few prior studies have investigated the uncertainty quantification of ML models [ 12 , 38 ], their uncertainty estimates are only indicative of prediction errors but do not translate to error-controlled predictions. In contrast, CPEC enables researchers to specify a maximum level of error rate and produces a set of predictions whose error rate is guaranteed to be lower than the specified level. Additionally, CPEC stands out by providing risk estimates, which delivers insights into the potential outcomes even before experimental validation and aids in the strategic allocation of experimental resources. One limitation of the CPEC framework is that when under covariate shift (i.e., P calib ( X ) ≠ P test ( X )), the data assumption of CPEC that the data in the calibration and test sets are i.i.d. is violated, which might lead to suboptimal FDR control performances ( S6 and S7 Figs). Although weighted conformal prediction frameworks have been proposed to address this limitation [ 39 ], the quantification and control of non-monotonic risk functions (e.g., FDR) under covariate shift remained a challenging problem. In this work, we define the error rate as the false discovery rate (FDR) to reflect the practical consideration in experiments where the goal is to maximize the success rate of hypothesis validation given a limited test budget. Nevertheless, the CPEC framework can be extended to support other forms of error rates, such as false negative rate [ 13 ]. In addition to protein function annotation, we expect CPEC to be a valuable tool for researchers in other biological discovery applications particularly when a balance between the experimental budget and the high yield rate is desired, such as drug target identification [ 40 ], material discovery [ 41 ], and virtual molecule screening [ 38 ].
Supporting information
S1 text. supplementary information..
Additional methodology, detailed experiment descriptions, and further evaluation experiments are included in the file.
https://doi.org/10.1371/journal.pcbi.1012135.s001
S1 Fig. Performance evaluation of representative baseline methods for EC number prediction.
We evaluated DeepEC, ProteInfer, DeepFRI, and CPEC (PenLight2) for predicting the 4th level EC number, using sample-averaged precision and recall as the metrics. DeepEC and DeepFRI were evaluated using the only trained model provided in their repositories, whereas ProteInfer was assessed using 5 different trained models. DeepFRI was trained on the same dataset as PenLight2 while DeepEC and ProteInfer were trained by their respective datasets. PenLight2 was trained using 5 different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s002
S2 Fig. CPEC achieves FDR control for EC number prediction.
Platt scaling [ 34 ], RED [ 36 ], and Monte Carlo dropout [ 35 ] were further evaluated as thresholding strategies, in comparison to CPEC. Due to the requirements of the methods, RED and MC dropout were applied on top of an MLP model. The results of CPEC and all of the thresholding strategies were averaged over five different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s003
S3 Fig. Performance evaluation of representative baseline methods for EC number prediction on test proteins with [0, 30%) sequence identities to training proteins.
We evaluated DeepEC, ProteInfer, DeepFRI, and CPEC (PenLight2) for predicting the 4th level EC number, using sample-averaged precision, recall, F1 score, nDCG, and coverage as the metrics. DeepEC and DeepFRI were evaluated using the only trained model provided in their repositories, whereas ProteInfer was assessed using 5 different trained models. DeepFRI was trained on the same dataset as PenLight2 while DeepEC and ProteInfer were trained by their respective datasets. PenLight2 was trained using 5 different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s004
S4 Fig. The FDR control of CPEC with different calibration set sizes.
The performances of CPEC’s FDR control were evaluated using calibration sets with various sizes (abbrev: calib. set size): 20%, 10%, 5%, and 1% of the total number of the training data. The same training data was used across all calibration set sizes to ensure consistency in the comparison. The black dotted line in the first panel represents the theoretical upper bound of FDR over test proteins. The results were averaged over five different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s005
S5 Fig. Performance evaluation of representative baseline methods for EC number prediction on test proteins that do not belong to the same CATH [ 37 ] superfamilies as the training proteins.
CPEC and three baseline methods (DeepEC, ProteInfer, and DeepFRI) were evaluated for predicting the 4th level EC number, using sample-averaged precision, recall, F1 score, nDCG, and coverage as the metrics. DeepEC and DeepFRI were evaluated using the only trained model provided in their repositories, whereas ProteInfer was assessed using 5 different trained models. DeepFRI was trained on the same dataset as CPEC, while DeepEC and ProteInfer were trained using their respective datasets. Training proteins not labeled in the CATH database were only removed from the training dataset of CPEC but not from the baseline methods’ training sets, which gave potential advantages to baseline methods. CPEC was trained using 5 different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s006
S6 Fig. Application of the FDR control for the EC number prediction of out-of-distribution (OOD) proteins.
The FDR control of CPEC was evaluated on a more challenging data split: no training and test proteins belong to the same CATH [ 37 ] superfamily. Training proteins not labeled in the CATH database were only removed from the training dataset of CPEC but not from the baseline methods’ training sets, which gave potential advantages to baseline methods. The results were averaged over five different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s007
S7 Fig. Application of the FDR control for the EC number prediction of out-of-distribution (OOD) proteins.
The FDR control of CPEC was evaluated on a more challenging data split: no training and test proteins belong to the same CATH [ 37 ] superfamily. A total number of 200 test proteins were sampled from the test set, and proteins that belong to the same superfamilies as the sampled test proteins were removed from the training set of CPEC. Training proteins not labeled in the CATH database were only removed from the training dataset of CPEC but not from the baseline methods’ training sets, which gave potential advantages to baseline methods. The results were averaged over five different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s008
S8 Fig. PenLight2, the base ML model of CPEC, outperforms the state-of-the-art methods for EC number prediction.
CPEC (PenLight2) and four baseline methods (including a baseline MLP model that takes the ESM-1b protein embeddings as the input) were evaluated for predicting the 4th-level EC number on more challenging test proteins with [0, 30%) sequence identities to the training proteins and the micro-averaged precision-recall curves were drawn. For each curve, the point with the maximum F1 score (Fmax) was labeled.
https://doi.org/10.1371/journal.pcbi.1012135.s009
S9 Fig. Evaluation of two point-uncertainty prediction approaches.
Two point-uncertainty prediction methods (Monte Carlo dropout (MC dropout) [ 35 ] and RED [ 36 ]) were evaluated in terms of uncertainty quantification. To make a fair comparison, a multi-layer perception taking ESM-1b protein embedding as the input was selected as the base ML model. The percentiles of the prediction variance (10th, 20th, 30th,…, and 100th percentiles) on the test set were used as the cutoffs. Predictions with variances larger than the cutoff were dropped. Observed false discovery rate (FDR), precision, recall, and coverage were used as metrics. The results were averaged over five different seeds.
https://doi.org/10.1371/journal.pcbi.1012135.s010
Acknowledgments
This work used the Delta GPU Supercomputer at NCSA of UIUC through allocation CIS230097 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program ( https://access-ci.org/ ), which is supported by NSF grants #2138259, #2138286, #2138307, #2137603, and #2138296. The authors acknowledge the computational resources provided by Microsoft Azure through the Cloud Hub program at GaTech IDEaS ( https://research.gatech.edu/energy/ideas ) and the Microsoft Accelerate Foundation Models Research (AFMR) program ( https://www.microsoft.com/en-us/research/collaboration/accelerating-foundation-models-research/ ).
- View Article
- PubMed/NCBI
- Google Scholar
- 13. Angelopoulos AN, Bates S. A gentle introduction to conformal prediction and distribution-free uncertainty quantification. arXiv preprint arXiv:210707511. 2021;.
- 14. Luo J, Luo Y. Contrastive learning of protein representations with graph neural networks for structural and functional annotations. In: PACIFIC SYMPOSIUM ON BIOCOMPUTING 2023: Kohala Coast, Hawaii, USA, 3–7 January 2023. World Scientific; 2022. p. 109–120.
- 18. Vovk V, Gammerman A, Shafer G. Algorithmic learning in a random world. Springer Science & Business Media; 2005.
- 19. Papadopoulos H, Proedrou K, Vovk V, Gammerman A. Inductive confidence machines for regression. In: European Conference on Machine Learning. Springer; 2002. p. 345–356.
- 21. Angelopoulos AN, Bates S, Fisch A, Lei L, Schuster T. Conformal risk control. arXiv preprint arXiv:220802814. 2022;.
- 22. Angelopoulos AN, Bates S, Candès EJ, Jordan MI, Lei L. Learn then test: Calibrating predictive algorithms to achieve risk control. arXiv preprint arXiv:211001052. 2021;.
- 23. Vovk V, Gammerman A, Saunders C. Machine-learning applications of algorithmic randomness. 1999;.
- 25. Hoeffding W. Probability inequalities for sums of bounded random variables. In: The collected works of Wassily Hoeffding. Springer; 1994. p. 409–426.
- 32. Brody S, Alon U, Yahav E. How attentive are graph attention networks? arXiv preprint arXiv:210514491. 2021;.
- 33. Wang Y, Wang L, Li Y, He D, Liu TY. A theoretical analysis of NDCG type ranking measures. In: Conference on learning theory. PMLR; 2013. p. 25–54.
- 35. Gal Y, Ghahramani Z. Dropout as a bayesian approximation: Representing model uncertainty in deep learning. In: international conference on machine learning. PMLR; 2016. p. 1050–1059.
- 36. Qiu X, Miikkulainen R. Detecting misclassification errors in neural networks with a gaussian process model. In: Proceedings of the AAAI Conference on Artificial Intelligence. vol. 36; 2022. p. 8017–8027.
3D Vessel Segmentation With Limited Guidance of 2D Structure-Agnostic Vessel Annotations
Ieee account.
- Change Username/Password
- Update Address
Purchase Details
- Payment Options
- Order History
- View Purchased Documents
Profile Information
- Communications Preferences
- Profession and Education
- Technical Interests
- US & Canada: +1 800 678 4333
- Worldwide: +1 732 981 0060
- Contact & Support
- About IEEE Xplore
- Accessibility
- Terms of Use
- Nondiscrimination Policy
- Privacy & Opting Out of Cookies
A not-for-profit organization, IEEE is the world's largest technical professional organization dedicated to advancing technology for the benefit of humanity. © Copyright 2024 IEEE - All rights reserved. Use of this web site signifies your agreement to the terms and conditions.
Increase Student Engagement and Outcomes with Hypothesis Social Annotation
Enhance student engagement, comprehension, and collaboration in your courses today with Hypothesis social annotation.
Hypothesis social annotation allows students and educators to collaboratively highlight, comment, and discuss any piece of online content directly in the margins.
Benefits of Hypothesis Social Annotation:
3-5x increase in student engagement.
Hypothesis significantly boosts student interaction with course materials.
Graduate-Level Comprehension for Undergraduates
Undergraduate students using Hypothesis comprehend course materials at a graduate student level.
18% Boost in Grades
Students can see an average 18% improvement in their grades when using Hypothesis.
32% Increase in Class Retention
Hypothesis helps increase class retention rates by 32%, keeping students engaged and enrolled.
Seamlessly Integrates in Your LMS
Hypothesis is easy to use and seamlessly integrated into your LMS for easy grading and annotation over course content.
Annotate Everywhere
Annotate over any piece of online course content including articles, webpages, YouTube Videos, JSTOR content, Bookshelf by VitalSource eTexts, and more!
Testimonials
Professor of English Cerritos College Francie Quaas-Berryman "I have found Hypothesis to be a great way to guide students through online reading, provide additional resources directly through the text, and engage them in online discussions of the text!"
ESOL Instructor Peralta Community College District Danitza Lopez "We love Hypothesis! This handy tool allows us to go deeper than any other platform because we can directly have discussions about a particular word/sentence/paragraph within a text. We even use it for peer reviews, and it works super well. It is such an easy tool to use, too!"
Professor of English Florida State University Candace Ward, Ph.D. "I’ve found Hypothesis invaluable in engaging students with assigned reading material…The students say very smart things; they engage with each other very civilly and respectfully, and they synthesize their annotations with other class discussions, projects, and readings."
Professor of Chemistry Rutgers University–Camden Dr. Georgia Arbuckle-Keil "I noticed that the personality of the students came through...I gain insight into the students beyond what is possible in the classroom. Even after we are back in person, I have enjoyed getting to know students via Hypothesis beyond a typical classroom response."
Associate Professor of Teaching/Instruction Temple University Marcia Bailey "Hypothesis is an excellent tool to engage students with the text and with each other. I used it almost weekly this semester."
Distinguished Service Professor of Anthropology Knox College Nancy Eberhardt, Ph.D. "Hypothesis gets students talking to each other about the reading outside of the classroom in a way that I have never been able to see happen before. They come to class much more ready to talk about the reading, and I have a sneak preview of the issues they found most compelling, as well as the ones they might have missed"
Distinguished Professor Information Science and Computer Science, University of Colorado Boulder Leysia Palen "With Hypothesis, everyone’s visible to each other and so we really do have a chance at creating a learning community. Students get to know each other’s names. Conversations with Hypothesis feel very warm and collegial. Nobody has to be afraid of raising their hand"
Proven Results:
Don’t just take our word for it, explore case studies from educators who have seen a significant impact on their courses:
Read Our Case Studies
Get Started
IMAGES
VIDEO
COMMENTS
Creating annotations. Log in to Hypothesis and use your cursor to select any text. The annotation adder will pop up, enabling you to choose whether to create a highlight (highlights are like private annotations with no related note) or to annotate the selected text. When creating an annotation, use the toolbar above your note to format text ...
Hypothesis is a free online tool designed to allow for collaborative annotation across the web. This guide will walk you through using Hypothesis with the Chrome extension. To use the bookmarklet in another browser, please refer to the hypothesis user guide for step-by-step instructions. It can be used to annotate web pages, PDFs and EPUB files ...
Open the text in Hypothesis as a student would and add your prompts as annotations. Consider these 10 different types of annotations. Use a text or a section of a text to model how you would engage with it. Open the text in Hypothesis and add annotations to share your own responses, analyses, and meta-comments.
Jeremy Dean, our Director of Education, introduces the Hypothesis annotation tool and shows how social reading can transform your classroom, making reading a...
Heather Staines, Hypothesis' Director of Partnerships, recreated this flash talk she gave at the STM Innovations Seminar 2017 in London.In just five minutes,...
Getting Started and Using Hypothesis in Canvas. Hypothesis, is accessed through Canvas Assignments [as an External] tool for both graded or ungraded activities. To get started and learn more see the following guides: Getting started with Hypothesis: Annotation tips and etiquette for students; An Illustrated Guide to Annotation Types
Hypothesis is a web annotation tool that allows users to annotate openly on websites, online journals, news articles, and blogs. It can also be used as a pri...
By default Hypothes.is is not activated in your Pressbook. In order to turn on the annotation tool, click Settings on the right-hand menu, then click Hypothesis. This will load the Hypothesis Settings Page. The Hypothesis Settings menu includes: Highlights on by default - check this box if you want the annotation highlights to be turned on by ...
Hypothesis allows authors to add additional insights about their work directly on the published article or book chapter. If made public by the author, the notes are visible to all users of Hypothesis, which creates a dynamic environment facilitating discussion and collaboration. Using Hypothesis not only allows authors to keep track of their ...
Hypothesis is a tool that lets you annotate readings with rich-text features, view annotations added by your instructor, and respond to other classmates' annotations. This guide will teach you how to use Hypothesis. Procedure. 1. In the Blackboard course with the Hypothesis assignment, click on the Hypothesis assignment to open it, then click ...
Hypothesis is a social annotation tool that quickly allows you to make class readings more active, visible and social.For more information on social annotation, see Annotation 101 and Back to School with Annotation: 10 Ways to Annotate with Students. Annotation can help students with reading comprehension and developing critical thinking about course materials.
Using multimedia & tags in annotations; Using Hypothesis with small groups; Creative ways to use social annotation in your course; Show-and-tell participatory workshop; Liquid Margins. Hypothesis hosts a recurring web "show" featuring instructors and staff to talk about collaborative annotation, social learning, and other ways to make ...
If you just do exam 1, you filter for everything the class tagged exam 1. You can then click the contextual link by the annotation, and hypothes.is will open that page, navigate to the highlighted text and display the annotation. (Note: In LibreText the highlights are hidden by default, and you need to click the "eye" with the slash over it ...
A different tool, Perusall, allows students to annotate e-books/e-textbooks, although the selection of texts is limited. Hypothesis allows editing in public, in private groups, or privately. I use a private group to create a class-specific space where we can engage with each other without having to filter out comments left by others.
Getting started. Now you have the extension up and running. It's time to start annotating some documents. Create an account using the sidebar on the right of the screen. Pin the Hypothesis extension in Chrome (1 and 2), then activate the sidebar by clicking the button in the location bar (3). Go forth and annotate!
Hypothes.is is an open source tool/plugin for social, collaborative asynchronous annotation. Collaborative annotation is an effective methodology that increases student participation and engagement, expands reading comprehension, and builds critical-thinking skills and community in class. Annotation can also increase faculty, student and ...
Hypothesis is an open platform for annotation and discussion of web resources. Cambridge is partnering with Hypothesis to enable authors, editors and readers to annotate and discuss the research we publish on our platform, Cambridge Core. You can use the Hypothesis annotation tool on Cambridge Core for the following content: Please find how to ...
You can then click the contextual link by the annotation, and hypothes.is will open that page, navigate to the highlighted text and display the annotation. (Note: In LibreText the highlights are hidden by default, and you need to click the "eye" with the slash over it (figure 2.4.2 2.4. 2.
Specifically, we use the function annotation problem of protein enzymes as an example to demonstrate our method. The underlying computational problem of function annotation is a multi-class, multi-label classification problem as a protein can have multiple functions. ... For each null hypothesis , we calculate a corresponding p-value p i: ...
Here, we demonstrate that large language models (LLMs) can accurately. 22 identify genes likely to be causal at loci from GWAS. By evaluating the performance of GPT-3.5. 23 and GPT-4 on datasets of GWAS loci with high-confidence causal gene annotations, we show.
To avoid the repetitive and costly annotating process for each vascular structure and make full use of existing annotations, this paper proposes a novel 3D shape-guided local discrimination (3D-SLD) model for 3D vascular segmentation under limited guidance from public 2D vessel annotations. The primary hypothesis is that 3D vessels are composed ...
Each token in the hypothesis was a button in the annotation interface and annotators wereinstructed toclickonincorrect tokenstomark errors. Unmarked tokens were assumed OK. ... Hypothesis: session .use only cookies bestimmt , ob das Modul nur <bad> mit </bad> Cookies die Session-ID auf dem <bad> Client-Betriebssystem speichert </bad> .
The expansion and contraction of the A. dimidiatus gene family were identified using the phylogenetic tree constructed by the single-copy homologous gene set, along with the estimated species divergence time. Furthermore, gene family selection pressure analysis was conducted using all single-copy orthologous genes identified in the eight species.
Enhance student engagement, comprehension, and collaboration in your courses today with Hypothesis social annotation. Hypothesis social annotation allows students and educators to collaboratively highlight, comment, and discuss any piece of online content directly in the margins. Start Enhancing Your Learning Experience Today!