Synthesis of Drugs ¶
Thomas Mavromoustakos (National Hellenic Research Foundation), Claude Cohen (Synergix), Elie Cohen (Synergix)
By creating new molecules and synthesizing new drug candidates, chemistry plays a central role in drug discovery and development. This chapter introduces the strategies and tactics used in organic synthesis. Concepts such as retrosynthesis, reaction classifications, protecting groups, racemic or asymmetric syntheses and directed organic syntheses are described and illustrated with examples. The syntheses of some common drugs are also presented. The chapter reviews some of the computer programs available in automated synthesis planning and current databases used in organic synthesis.
Number of Pages: 198 (±4 hours read)
Last Modified: September 2005
Prerequisites: None

Introduction ¶
Why to synthesize a new molecule ¶.
Synthetic organic chemistry is a powerful field that governs much of drug discovery and development. It enables us access to new chemical compounds such as analogs of natural products or novel synthetic structures that can be used as probes to understand biological events and the derivation of useful therapeutic agents. It also plays a pivotal role in the preparation of the necessary quantities for clinical trials and bulk production of the drug when it becomes commercialized.

Douglas S. Johnson and Jie Jack Li
John Wiley and Sons, Hoboken 2007
Drug Discovery ¶
The synthesis of new molecules plays a key role in the drug discovery process. A drug candidate results from a lengthy and intense research activity where many molecules are synthesized and extensively tested. This generates informative structure-activity relationships for the conversion of this knowledge in the optimization and development of the drug candidate.

Bulk Production ¶
The manufacturing and bulk production of a drug is costly and involves heavy industrial installations. Millions of dollars can be saved by reducing the production costs of a drug. For example, for more than 60 years the commercial synthesis of Vitamin C has involved a six-step chemical process. Recently, an intelligent modification of the synthetic process has shortened it to only three major steps. More than 110,000 tons of Vitamin C are produced annually; the economical and ecological impact on what has become a $1 billion-plus industry are considerable.

Goal of the Synthesis ¶
The aim of the synthesis is to prepare a target compound (TC, the drug candidate) which is synthesized by converting the starting material (SM) after a series of chemical interconversions have been carried out. A, B ... are intermediate products, whereas X, Ψ, Ω ... represent the reagents and conditions of the chemical reactions.

General Requirements before Starting ¶
Before starting an organic drug synthesis, the requirements are generally well established. The target compound should be of low molecular weight. We need cheap, non toxic, readily available materials and keep the synthesis short. Whenever possible, stereospecific reactions with high yields must be planned. The reactions should be adaptable to large-scale production methods with intermediate products that are easy to isolate, purify and identify.

Number of Steps and Intermediates ¶
Drug synthesis is a multiple step process. The strategy consists of moving from SM to TC with a minimum number of steps. It is extremely important to address the issue of reducing the number of steps: in drug discovery this accelerates the discovery cycle, and in process manufacturing this reduces production costs of the drug. For example, nifedipine is synthesized in 2 steps, viagra and zyvox in 9, lipitor and cephalexin in more than 11 steps.

Measurable Reaction's Characteristics ¶
Many characteristics of a reaction can be observed and measured as the reaction proceeds. These typically include the yield, the reaction rate, the intermediates formed and the products obtained. Some of these are discussed in the following pages.

Yield ¶
The yield of a reaction is a percentage that indicates the amount of product actually obtained as compared to the theoretical yield (which is the amount of product predicted to form on the basis of the balanced chemical equation). A yield of 80% is good per step. Note that a five step reaction with an average yield of 80% for each step would give an overall yield of only 33%: this shows that overall yields drop off rapidly even with high yields at each stage. It may be possible to increase the effectiveness of a given step by changing the experimental conditions such as the reagents, the temperature, the time or the catalysts.

Reaction Rate ¶
Chemical reactions occur at different rates. Some reactions are extremely fast while others are very slow, depending on the nature of the reactants and products and the conditions under which the reaction takes place. Factors that affect reaction rates include temperature, the concentration of the reactants, the pH, the physical state of reactants, the presence of a catalyst and light.
Product Selectivity ¶
The result of a reaction between a reactant and a reagent is not always a unique product; multiple competing reactions can take place and produce different products. Normally for the synthesis of a drug, only one of the products is needed and the reaction is optimized to maximize the formation of the desired compound as far as possible.

Regioselectivity and Regiospecificity ¶
Above we saw that a reaction can yield different products, and these products are not isomers . However, there are reactions that produce isomeric products, such as regioselective reactions. A simple example is illustrated below, where the reaction of HBr with propene gives 1-bromopropane and 2-bromopropane (if this regioselective reaction had given only one product it would have been called "regiospecific").
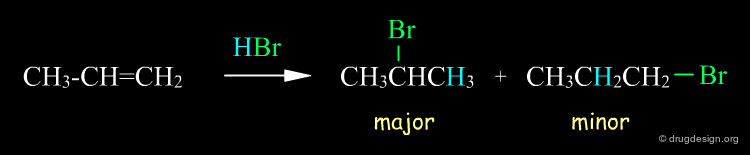
Stereoselectivity and Stereospecificity ¶
A reaction is stereoselective when there is preferential formation of one stereoisomer over another. A reaction is sometimes termed stereospecific when there is very high stereoselectivity. Stereoselectivity is an important variable in the synthesis of many drugs.

Thermodynamic and Kinetic Properties of the Reaction ¶
Thermodynamics and kinetics control the course of every chemical reaction. A reaction can be thermodynamically controlled (when the product composition is governed by the equilibrium thermodynamics of the system), or kinetically controlled (when the product composition is governed by competing rates of formation of products), in which case the formation of the product with the lowest activation energy is favored.

Thermodynamics ¶
The enolization of keto-3 steroids is an example of a thermodynamically controlled reaction. The composition of the two enols obtained is dictated by their relative energies: there is a difference of 2.4 kJ/mol, in favor of the Δ 2 enol.

249. Steroids. Part CLXXI. Factors Controlling the Direction of Enol Acetylation of 3-oxo-steroids Berkoz B, Chavez EP and Djerassi C J. Chem. Soc.
The Torsion Angle Concept in Conformational Analysis Bucourt R Topics in Stereochemistry 8 1974
Kinetics ¶
The nucleophilic attack of a ketone in a chiral environment is under kinetic control: unequal amounts of enantiomeric alcohols are produced because the two diastereoisomeric transition states do not have the same energy.

Determinants of a Chemical Reaction ¶
Thermodynamic and kinetic properties determine the course of a chemical reaction. The reaction is driven by the energies of the reagents and products (thermodynamics) and of the transition states (kinetics). Steric, electronic and solvent effects for instance contribute to the energies of such molecular systems.

Steric Effects ¶
Steric effects can influence conformational preferences or stabilizing transition states of reactions. They control the course of the regioselectivity of many reactions such as additions, eliminations, substitutions or rearrangements. For example the regioselective formation of the less crowded alkene in the following Hofmann elimination is due to steric effects resulting in the low energy of the corresponding transition state.

Electronics Effects ¶
Electronic effects can influence the resonance, the inductive effects, the stereoelectronic features, the basicity of nucleophiles, the acidity of electrophiles, the stabilization of ionic structures etc... The classical case is the ortho, meta or para substitutions in Friedel Crafts reactions. In the example below we see how the rate of N-Nitrosation of N-Benzylpivalamides can be accelerated by para electron-donating groups.
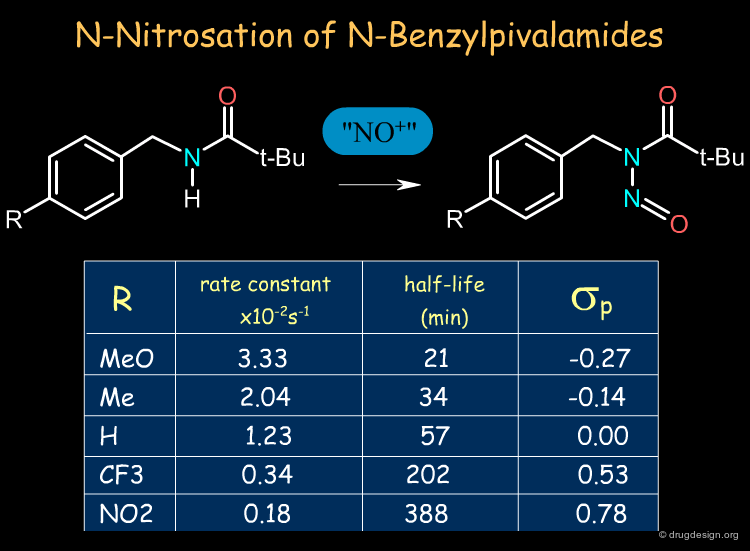
Electronic Effects in the N-Nitrosation of N-Benzylpivalamides Ron W. Darbeau, Rebecca S. Pease, and Edson V. Perez J. Org. Chem. 67 2002
Solvent Effects ¶
The solvent performs the mechanical but vital role of allowing immiscible reactants to come together rapidly. The example shown below illustrates the influence of the solvent in a Still-Wittig rearrangement yielding opposite stereoselectivities depending on the nature of the solvent.
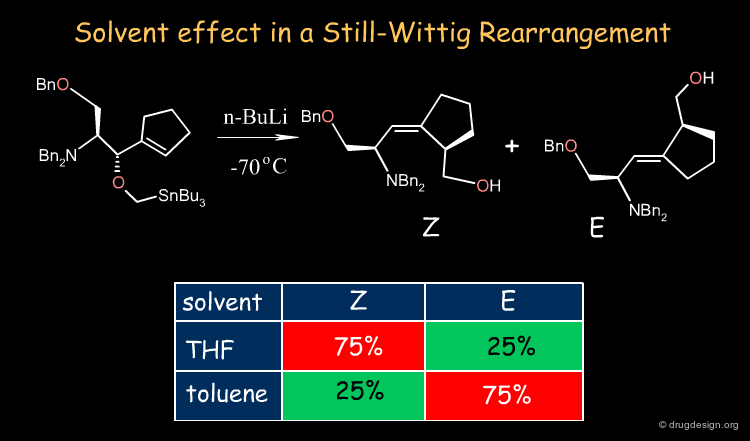
Solvent-Dependent Stereoselectivity in a Still-Wittig Rearrangement: An Experimental and ab Initio Study Scott A. Hart, Carl O. Trindle, and Felicia A. Etzkorn Organic Letters 3 2001
How to Influence a Reaction? ¶
Because of their complexity, the chemist is less concerned with the energies of the products or the transition states, and is more interested in practical ways of influencing the reaction so it can be oriented towards the desired end products. The concrete parameters that the chemist can influence are for example the choice of the reactants, reagents, concentrations, temperature, catalyst, pH, solvent, etc...

Reactant Choice ¶
Once a reaction has been selected it can be influenced by changing various experimental parameters - first and foremost, the choice of the reactants and reagents. In the example below (molecules that are important in the discovery of EGF-R kinase inhibitors ) the nature of the halogen in position 6 proved to be critical for enabling the reaction to take place. A fluorine atom in position 6 is easily displaced by primary and secondary amines, whereas chlorine in the same position cannot be displaced by amines or other nucleophiles.
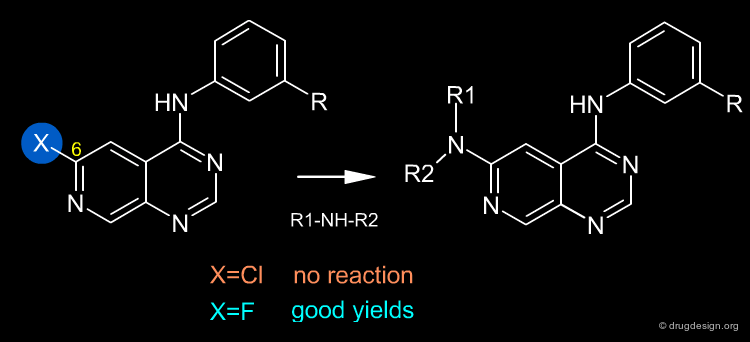
Synthesis of 4-(Phenylamino)pyrimidine Derivatives as ATP-Competitive Protein Kinase Inhibitors with Potential for Cancer Chemotherapy Rewcastle G.W.; Denny W.A.; Showalter H.D.H. Current Organic Chemistry 4 July 2000
Reagent Choice ¶
The example below illustrates how it is possible to direct the stereoselectivity of a reaction by modifying the reagent. It shows how it is possible to influence the course of the reaction towards the desired product with the right choice of the nucleophile.

Reaction Conditions ¶
Once a specific reaction has been selected, it is implemented - which is one of the chemist's most time consuming jobs. A whole set of conditions need to be defined: the reagents, the time, the temperature, the concentration or the physical state of the reactants, the solvent, the pH, the catalysts and their combinations to obtain the desired product. In the example below, 50 trials were carried out before the best set of conditions (with a yield of 96%) was found. In unfavorable conditions, the reaction does not work well and cannot be rectified.

Influence of pH ¶
The following example illustrates how a Friedel-Crafts acylation in water can be sensitive to pH. At pH 7.5 the indolic system is efficiently acylated with ethyl glyoxylate. At this pH the yields are as high as 95%; they drop to 17% at pH 11.3.

Friedel-Crafts reactions in water of carbonyl compounds with heteroaromatic compounds Wei Zhuang and Karl Anker Joergensen Chemical Communications 13 2002
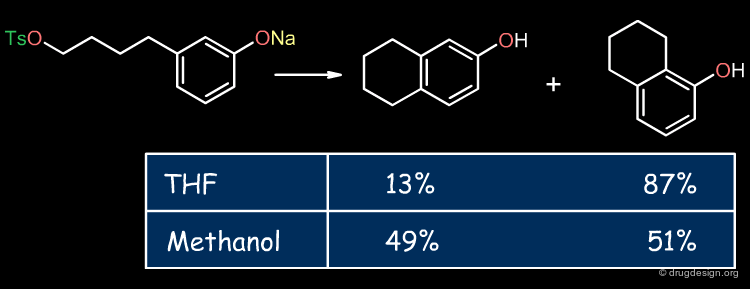
Influence of the Solvent ¶
The solvent can substantially affect the solvation of reactants, products or transition states resulting in variations in the regioselectivity, reaction rate or equilibrium of a reaction. From solvent to solvent the solvation energies may vary by up to 40 kJ/mol.
Catalysts ¶
The role of the catalyst is to facilitate the formation of the desired product by reducing the activation energy of the reaction. Over the last few decades chemists have devised a vast range of chiral and non-chiral catalysts that contain complex inorganic, organic and biological catalysts that are designed for specific reactions such as Suzuki coupling, asymmetric metathesis etc....

Tools for Following the Progression of a Reaction ¶
Because a reaction takes place in many steps, it is important to have the right tools to make sure that the compounds obtained at each step are the expected ones. Did the reaction take place? Was the reaction complete? How many products are in the flask? What is the main product? These are some of the questions which need to be answered. There is a broad range of analytical tools available to monitor the progression of a chemical reaction. Spectroscopy and chromatography are the most indispensable means used in all laboratories. It is possible to rely on a single analytical method or in a combination or several of them.

Spectroscopy ¶
Spectroscopy is an important tool to test for the correct progression of the synthesis of a drug. A spectrum can be obtained (absorption or emission), that provides a fingerprint that is characteristic of the molecular structure concerned. It can be compared to the spectrum of a known compound, or analyzed in terms of the chemical structure of the compound considered. Analytical spectroscopic tools include mass spectroscopy, infrared, nuclear magnetic resonance , ultraviolet, circular dichroism and X-ray crystallography.

D. Williams and I. Fleming
McGraw-Hill 1989
Mass Spectra (MS) ¶
In mass spectrometry, a substance is bombarded with an electron beam with sufficient energy to fragment the molecule. The ions generated are accelerated through a magnetic field and are sorted on the basis of a mass-to-charge ratio. A precise molecular mass is obtained and the analysis of the spectrum involves re-assembling the fragments to generate the exact structure of the original molecule.

Infrared (IR) ¶
Absorption of infrared radiation results in the excitation of vibrational, rotational and bending modes of groups of atoms, while the molecule itself remains in its electronic ground state. The functional groups present in the molecule can easily be identified through their characteristic absorption wavelengths.

Nuclear Magnetic Resonance ( NMR ) ¶
Nuclear magnetic resonance is a spectroscopic technique that provides information about the number and types of atoms in a molecule. NMR spectrometers are tuned to certain nuclei such as 1 H, 13 C, 19 F and 31 P. Absorption in the low-energy radio-frequency part of the spectrum causes excitation of nuclear spin states; high-resolution spectroscopy distinguishes atoms in different locations in the molecule.

Ultraviolet (UV) ¶
Absorption or emission of ultraviolet or visible light by a molecule depends on electronic transitions between molecular orbital energy levels in the molecule. UV spectra can be used to identify an unknown compound by comparative analyses. It is not used to identify the presence of functional groups, but rather to show relationships as conjugation. UV spectra may be also used to determine the concentration of a compound in a mixture.

Circular Dichroism (CD) and Optical Rotatory Dispersion (ORD) ¶
Circular dichroism is used for the spectroscopic analysis of chiral molecules by measuring differences in the absorption of left-handed polarized light versus right-handed polarized light, which arise from the structural asymmetry of the chiral moieties .

X-rays ¶
Solid state X-ray crystallography is used to reveal the structure of a compound, information that could be invaluable when its 2D formula is problematic. For example, Dorothy Hodgkin and her team at Oxford were able to demonstrate the structure of penicillin by the resolution of its X-ray diffraction data. Dorothy Hodgkin was awarded the Nobel Prize in chemistry (in 1964) for this achievement that enabled chemists to develop synthetic ways to make this antibiotic - and many related analogs .

Chromatography ¶
Analytical chromatography is essential for following the progression of a synthesis. Based on differential migration of solutes, it can identify reagents, reactants and products that are present in the reaction mixture (by comparing it with the corresponding pure compounds). The technique was developed in the 1940s with paper chromatography, and in the 1950s with gas (GC) and thin layer chromatography (TLC). Today modern chromatography has generated a broad spectrum of techniques including high performance liquid chromatography (HPLC) for both analytical and preparative purposes.

Actelion Pharmaceuticals Ltd., Reproduced from Actelion, with permission Actelion
High Performance Liquid Chromatography (HPLC) ¶
High performance liquid chromatography is a tool for quantifying and analyzing mixtures of chemical compounds. The method was developed in the mid-1970s and has become the most popular method of analysis because it is easy to use.

Chiral Chromatography ¶
The separation of enantiomers by chiral HPLC is a useful method for the analysis of chiral substances. It can be a very efficient method for the separation of racemic compounds, to control the optical purity and also for the preparation of optically pure drugs.

Design Strategy ¶
Strategy like a general in the battle ¶.
The synthesis of the target compound can be viewed as a battle in which the strategy that is best suited to achieving the upcoming goal is applied. The current status must be permanently assessed and the strategy revised. The medicinal chemist must find the appropriate reagents at minimum price and the best synthetic scheme to achieve this goal as quickly as possible.

Flexibility in the Strategy ¶
Many syntheses contain surprises and the chemist must be prepared to circumvent difficulties. "Flexibility" is the keyword and must be present at multiple levels, as discussed in the following pages.

Flexibility in the Synthetic Program ¶
In terms of chemistry, the synthetic scheme must be as flexible as possible. As many problems as possible must be predicted in advance at the design stages of a chemical program, so that there are alternative solutions available if a reaction fails to work.

Flexibility in the Target Molecule ¶
Flexibility should be uppermost in the chemist's mind. After all a chemist "synthesizes" an idea rather than a molecule... In chemistry if the result diverges from the expected outcome, you need to return to the hypothesis. Based on accumulated knowledge and imagination, another molecule must be designed that fits the working hypotheses better than the original one.

Nicolaou Statement on the Trip to Ithaca ¶
K.C. Nicolaou use to say says that organic synthesis is like the journey from Troy to Ithaca: this trip is full of adventure and new discoveries; encounters with Laistrygonians and Cyclops... For the medicinal chemist a second journey begins when the molecule enters the cycle of biological testing...

Linear and Convergent Strategy ¶
The synthesis of the molecule is like putting together a jigsaw puzzle. You can assemble complementary blocks, or you can make certain regions independently and then connect them. In organic synthesis, these two approaches are known as the linear and convergent strategies.

Linear Strategy ¶
In a linear strategy the molecule is constructed in a stepwise fashion, following a linear reaction sequence. The global yield of a linear synthesis decreases exponentially with the number of steps. Brevetoxine can be synthesized in 83 steps with an average yield of 91% per step, however the global yield is only 0.043% !

Example: Captopril Linear Synthesis ¶
Captopril can be synthesized in four steps according to the linear synthesis shown below, that starts from 2-methylacrylic acid.

Design of specific inhibitors of angiotensin converting enzyme: new class of orally active antihypertensive agents. Ondetti MA, Rubin B and Cushman DV. Science 196 1977
Convergent Strategy ¶
In the convergent synthesis two or more direct precursors of a target are constructed independently and then assembled in the last step of the synthesis.

Convergent Advantage ¶
Convergent routes present advantages over linear approaches with respect to speed, time, yields and manpower. Furthermore they have greater flexibility for producing series of analogs . The impact of low yield reactions is less dramatic in a convergent than in a linear approach, as illustrated in the theoretical examples below.

Example: Losartan Convergent Synthesis ¶
Losartan can be synthesized in a convergent manner as shown below. The imidazole and biphenyl precursors 4 and 8 are first prepared separately from (1 and 2) and (5 and 6), respectively, and they react together in one of the last steps of the synthesis to give the cyano analog 9, which is then transformed into the desired losartan drug 10 with the desired tetrazole moiety .

Nonpeptide angiotensin II receptor antagonists: the discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives David J. Carini, John V. Duncia, Paul E. Aldrich, Andrew T. Chiu, Alexander L. Johnson, Michael E. Pierce, William A. Price, Joseph B. Santella, III, Gregory J. Wells, and et al. J.Med.Chem.
Nonpeptide Angiotensin II Receptor Antagonists: The Next Generation in Antihypertensive Therapy Ruth R. Wexler, William J. Greenlee, John D. Irvin, Michael R. Goldberg, Kristine Prendergast, Ronald D. Smith, and Pieter B. M. W. M. Timmermans J.Med.Chem. 39 1996
How to Analyze a Molecule for Synthesis? ¶
Molecules have their own identity and capabilities and the chemist assesses the molecular complexity of the target in terms of characteristics such as those indicated in the list below. However, the analyses depend entirely on the chemist's experience and creativity in combining chemical reactions and selecting the best of a set of possible routes to making the molecule.
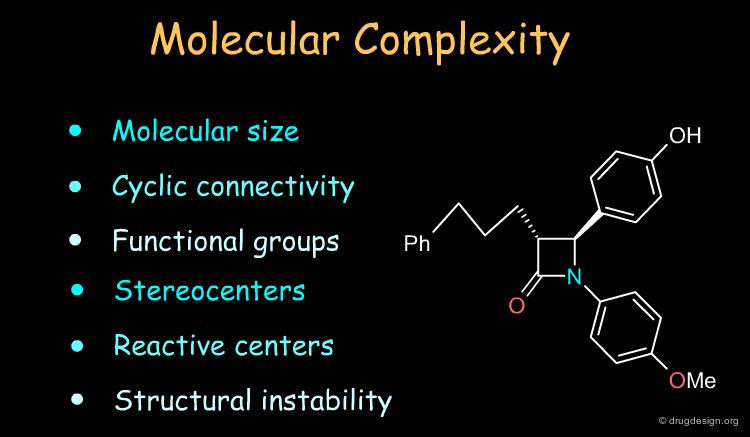
Three Methods for the Design of a Synthetic Program ¶
Having assessed the complexity of the target molecule, the next step is to find how to synthesize it. There are three basic methods: (1) adapt a known synthetic scheme; (2) use a building block strategy and (3) start from scratch.

Adapt Known Synthetic Schemes ¶
Adapting known synthetic schemes involves using available synthetical knowledge about other molecules. Rather than re-inventing the wheel, the chemist looks for similar molecules that have already been synthesized by other groups and decides whether the existing methods can be adapted for the synthesis of the target compound.

Literature and Patent Searches ¶
Chemical Abstracts, Beilstein, literature and patent searches provide useful ways for finding synthetic schemes that could be adapted for the preparation of the new molecule.

Database Searching with Computer Programs ¶
Today most information can be accessed through specialized databases. Database searching provides effective means for finding synthetic pathways and is a source of inspiration for the preparation of target compounds.

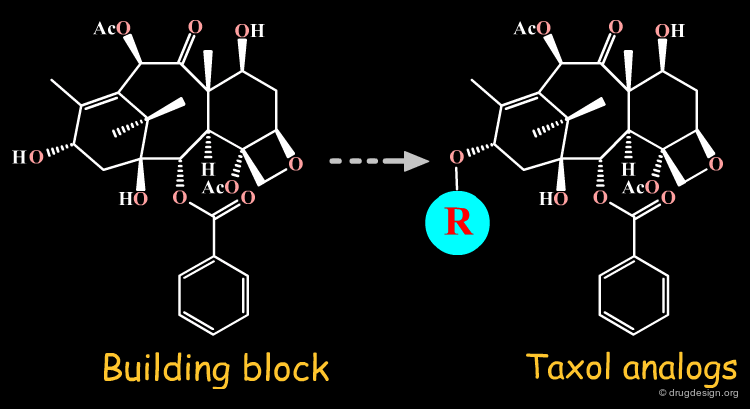
Consider a Building Block Strategy ¶
Unlike the existing knowledge adaptation of known synthetic schemes, the building block strategy aims at exploiting existing starting material. The building block strategy consists of incorporating elaborated and readily available fragments in the synthesis. The recognition of useful building blocks for a target molecule is of outmost importance because it can substantially simplify the chemical pathway.
Small Commercial Building Blocks ¶
The small building blocks for the synthesis of a novel drug must be commercially available and cheap. This is an issue that a medicinal chemist must constantly keep in mind.

Elaborated Building Blocks ¶
For semi-synthesis purposes, elaborated building blocks can be obtained from naturally occurring compounds. A great number of analogs can then be prepared with such scaffolds. Examples of moieties that can be either purchased or prepared are shown here in the taxol, codeine, staurosporine and penicillin families.
Starting from Scratch - Retrosynthetic Analysis ¶
When no analogy with a known molecule and no building block can be found, a synthetic route has to be invented 'from scratch'. Although the design is relatively straightforward for a simple structure, a method is needed when the molecule is complicated. "Retrosynthetic analysis" is an important method for analyzing and partitioning the problem. This is presented in the following pages.

Retrosynthetic Strategy ¶
In retrosynthetic analysis, a synthesis is planned by reasoning backward from the desired product. The approach was formalized by E.J. Corey (1990 Nobel Prize in Chemistry) by using standard rules. The steps are described using retrosynthetic arrows drawn as => and meaning "is synthesized from".

Computer assisted analysis of complex synthetic problems E. J. Corey Q. Rev. Chem. Soc. 25 1971
Towards the ideal synthesis P. A. Wender, S. T. Handy and D. L. Wright Chem. Ind. (London)
he Art and Science of Organic Natural Products Synthesis K. C. Nicolaou, E. J. Sorensen and N. Winssinger J. Chem. Educ. 75 1998
The Art and Science of Total Synthesis at the Dawn of the 21st Century K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran Angew. Chem. Int. Ed. 39 2000
E. J. Corey and X.-M. Cheng
Wiley, New York 1995
K. C. Nicolaou, Eric J. Sorensen
Wiley-VCH 1996
K. C. Nicolaou, Scott A. Snyder
Wiley-VCH 2003
Stuart Warren
John Wiley and Sons 1978
Cambridge Univ. 1982
Elsevier Vol 1: 1984; Vol.5:2004
Disconnection of Strategic Bonds ¶
In retrosynthesis the bonds of the target molecule are systematically disconnected to reveal the structures of precursor molecules (in orange in the figure), which are then examined in a similar way. One particularly useful tool in identifying good disconnections is to look at the relationship between functional groups. The key bonds that reveal the synthetic strategy are known as the "strategic bonds" and the reactive components of a synthetic step are called "synthons".

Strategic Bonds Revealed by Small Modifications ¶
The aim of retrosynthesis is to find disconnections and uncover strategic bonds that lead to simple starting material. Sometimes a disconnection can only be found if a small modification of a functional group is introduced into the molecule (in the example below, A was changed to A'). The most common transformations are: functional group addition (FGA), functional group interconversion (FGI) and functional group removal (FGR).

An example of functional group addition (FGA) is illustrated below. The transform introduced (addition of a methyl ester next to the carbonyl) cleverly increases the complexity of the target molecule: a Dieckmann reaction on the cyclobutane diester derivative leads to the desired structure.

An example with successive functional group interconversions (or interchanges) (FGI) is shown below. Finally, the target compound can be prepared from acetone as starting material.

An example of functional group removal (FGR) is shown below. The two phenyl groups are temporarily removed from the target structure, and are introduced in due time with a Grignard reaction. The synthesis is based on simple starting material such as ethyl acrylate, piperidine and bromobenzene (for the Grignard).

The Retrosynthetic Process ¶
The retrosynthetic process has often been compared to a game of chess. Organic synthesis is a science and a fine art in which masters such as Corey, introduced logical rules that are now the canon of every undergraduate synthetic chemistry course. The process starts by recognizing the key functional groups of the target compound and their possible transformations (e.g. FGA, FGI and FGR). Strategic bonds are then disconnected and assessed to see whether they lead to simple starting material.

From TC to SM ¶
Disconnecting in the middle of the molecule or at branch points from chains may yield useful simplifications. After locating the functional groups and their relationships all C-X and C-C bonds are then systematically disconnected, until available starting material is found. Retrosynthetic schemes provide useful material for thinking about ways precursors can be prepared and whether they correspond to reliable reactions. The chemistry associated to a disconnection is usually obvious; however in the intermediate steps the transformations of precursors may require more creativity.

Development of a Scalable Process for an Endothelin Antagonist, CI-1034 Thomas E. Jacks 25th Symposium on Synthetic Organic Chemistry, Holland, Michigan
The Synthetic Program ¶
As long as a retrosynthetic pathway does not end up with accessible starting material, the analysis is worth nothing. The most promising scheme finally adopted represents "the project": it serves to define phases, milestones and work plans for the group of chemists involved in the synthetic program.

The Tetrodotoxin Example ¶
The tetrodotoxin example is illustrated here; it shows the retrosynthetic scheme adopted by the Kishi group, and how the synthesis was developed.

Synthetic studies on tetrodotoxin and related compounds. III. Stereospecific synthesis of an equivalent of acetylated tetrodamine Y. Kishi, M. Aratani, T. Fukuyama, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura, and H. Kakoi J. Am. Chem. Soc. 94 1972
Synthetic studies on tetrodotoxin and related compounds. IV. Stereospecific total syntheses of DL-tetrodotoxin Y. Kishi, T. Fukuyama, M. Aratani, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura, and H. Kakoi J. Am. Chem. Soc. 94 1972
Simple Exercise in Retrosynthesis ¶
Suppose we want to synthesize the compound shown below, and in our laboratory we only have ethanol, acetone and all the inorganic reagents available. How do we proceed?

Retrosynthesis ¶
Two retrosynthetic routes are feasible. We can use the reagents CH 3 COCH 3 and CH 3 CH 2 MgCl, or, alternatively, CH 3 COCH 2 CH 3 and CH 3 MgCl. We choose the first option because butanone is not available in the laboratory.
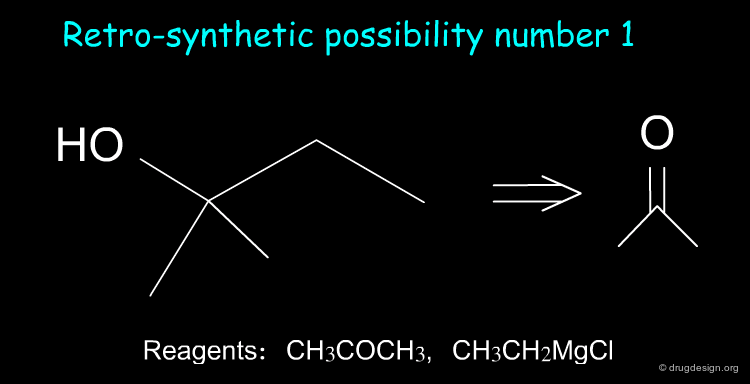
Synthesis ¶
The synthesis based on the chosen retrosynthetic route adopted can then be implemented. Starting with ethanol the desired compound is obtained as follows.

Disconnection Methods for Retrosynthesis ¶
The disconnection method plays a central role in retrosynthetic analyses. For a drug to be prepared, there are three ways to break or make a bond which can be used in retrosynthetic analyses. These are known as the homolytic, the heterolytic and the pericyclic disconnections.

Homolytic Disconnection ¶
Homolytic fission is a free radical disconnection. Free radical disconnections are generally not very useful in the synthesis of novel drugs because the reconnection reaction proceeds through free radical mechanisms, which usually produce mixtures and are not desirable in the synthesis of a drug.

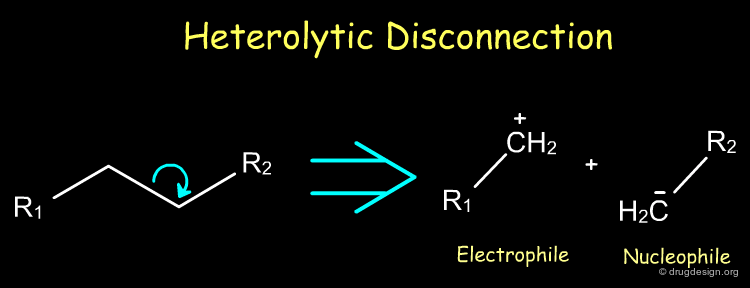
Heterolytic Disconnection ¶
Heterolytic disconnections are very useful because they result in the formation of electrophile and nucleophile species. Such species are synthons that can be converted into a reagent for the initiation or the propagation of a synthetic scheme.
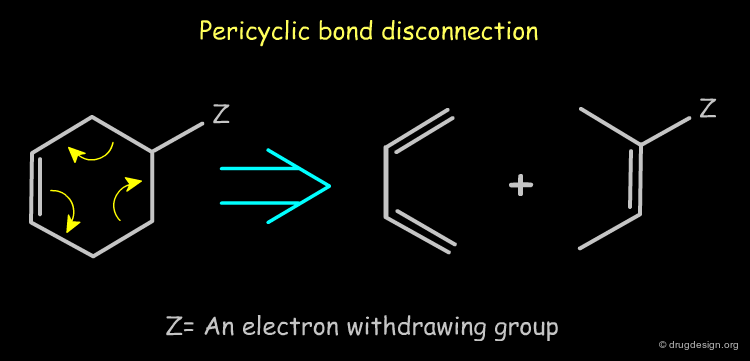
Pericyclic Bond Disconnection ¶
Rings that carry an electron-withdrawing group can be disconnected by a reverse pericyclic mechanism (i.e. Diels-Alder). Such disconnections are useful in synthetic medicinal chemistry because they lead to synthons that can be converted to reagents.
Combining the Three Synthetic Methods ¶
A synthesis can be based on existing knowledge, on existing material, or on creative design. A good synthetic route is generally an intelligent combination of all three, incorporating known and reliable chemical reactions, readily available compounds, and a good dose of ingenuity.
Synthetic Tactic ¶
Principles of synthetic tactics ¶.
Once the global strategy has been defined, a plan of attack is formulated, which defines specific actions to achieve the goal. These actions consist of a set of maneuvers, each of which requires adequate available resources to achieve the ultimate goal.

Reaction Classifications ¶
One of the best ways to organize a plan of attack is to use operational classifications of chemical reactions. There are different ways of characterizing organic reactions, which are useful tools for recognizing patterns of chemical reactivity. These are crucial to the design of a reaction. Some of these classifications are presented in the following pages.

By Changes Occurring in the Reactant Molecules ¶
When considering the structural changes occurring in the reactant molecules, it is useful to distinguish broad classes of reactions such as addition, elimination, substitution and rearrangements.

By Reaction Types ¶
It is sometimes useful to describe reactions in terms of changes of properties (e.g. oxidation state, acidity etc...) or in terms of species that seek out charged centers (e.g. nucleophiles, electrophiles). In the example below, we look at the oxidation changes of the carbon atoms of the double bond: one has been oxidized (C2) and the other (C3) reduced. Note that this type of description is independent of the mechanism.
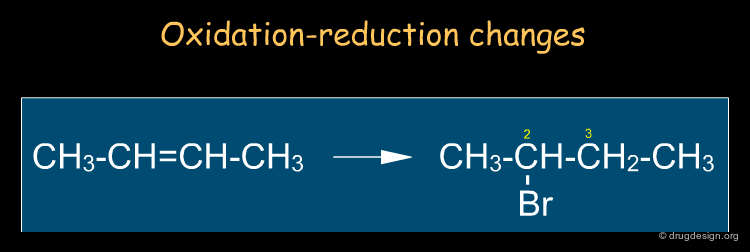
By Functional Groups ¶
Functional groups characterize the reactivity of molecules. Since most reactions occur at, or adjacent to functional groups, it is useful to identify them in terms of chemical functionality. Below are given some examples of functional groups with their most characteristic reactions.

Protecting Groups ¶
The design of a synthetic pathway often requires a reaction to be carried out at a certain center of the molecule. However, another center may interfere and undergo a similar undesired reaction. The synthetic medicinal chemist must find a way to protect the one center and allow the reaction to occur at the other one. For example if the dipeptide Phe-Gly is needed, it can be prepared by the condensation of the two amino-acids Phe and Gly; however if these amino-acids are not properly protected, the reaction can produce a mixture of the Phe-Gly, Phe-Phe, Gly-Phe and Gly-Gly dipeptides.

Protecting Reactive Centers ¶
Protecting groups are necessary to protect a reactive center that at a certain step should not be modified. Their role is to mask the particular center in order to allow a transformation to be carried out elsewhere in a molecule. In our example, the Phe-Gly dipeptide can be obtained quantitatively, by protecting the amino of the phenylalanine (N-protection) and the carboxyl of the glycine (O-protection).

Requirements for Protecting Groups ¶
The protecting groups must have the following requirements: (a) easy to attach to the appropriate functional group; (b) form a stable structure that is not affected by the reaction conditions and reagents; (c) easy to remove once it is not required; (d) can be converted into another structure as part of the synthesis.
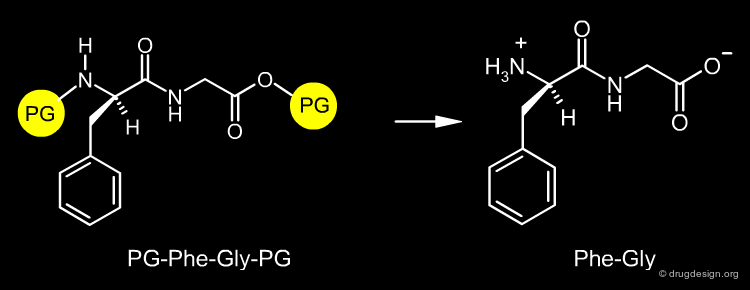
Protection and Deprotection of Amino Acids ¶
Amino groups are normally protected by converting them to amides whereas carboxyl groups are converted to esters. Examples of amino protecting groups are benzyloxycarbonyl (abbreviated as Z or Cbz) and tert-butyloxycarbonyl (Boc) that are introduced using the chloride. When needed, the protecting group can be easily removed: Cbz can be removed with catalytic hydrogenolysis under very mild conditions or HBr in acetic acid; and Boc can be removed with trifluoroacetic acid in methylene chloride or HBr in acetic acid.

Examples of Protecting Groups ¶
A variety of protecting groups for a certain functional group can be used and then removed at the intermediate or final steps of the synthesis. Some examples are presented in the following pages.

Alcohol Protection ¶
Several groups can be used for alcohol protection such as triphenylmethyl (trityl) acetate or trifluoroacetate. These groups may need different conditions for their removal: for example, trityl needs acid conditions while acetate or trifluoroacetate need base conditions.

Carboxylic Acid Protection ¶
The carboxylic group is one of the most common groups that must be protected during synthesis. Two examples of protecting groups and corresponding removal reagents are given below.

Amine Protection ¶
Amine groups are often present and need to be protected properly. In particular peptide synthesis requires their selective protection and removal.

Phenol Protection ¶
The hydroxyl group of alcohols and phenols frequently requires protection. The benzyl group is a suitable protecting group which can be removed by catalytic hydrogenation.

How to Assess the Quality of a Synthesis? ¶
There is no single correct pathway to the synthesis of a molecule. Some routes, however, are better than others. Measurable means for assessing the quality of a synthesis exist and include: the number of steps, good selectivity, good yields, economy of raw material and mild reaction conditions. Other factors are difficult to quantify but are also important, such as the creativity and elegance, practicality and convenience of the steps, flexibility with respect to problems, lack of involvement of toxic compounds or laborious purification procedures.

Syntheses of Swainsonine ¶
Swainsonine illustrates how a molecule can be synthesized by many different routes. Look at the corresponding articles and rank them in terms of your own preference.

Asymmetric synthesis of (-)-swainsonine, (+)-1,2-di-epi-swainsonine, and (+)-1,2,8 -tri-epi-swainsonine. Lindsay KB, Pyne SG. J Org Chem. 67(22) 2002
Synthesis of the new mannosidase inhibitors , diversity-oriented 5-substituted swainsonine analogues, via stereoselective Mannich reaction. Fujita T, Nagasawa H, Uto Y, Hashimoto T, Asakawa Y, Hori H. Org Lett. 6(5) 2004
Stereoselective synthesis of swainsonines from pyridines Gerres Heimgaertner, Dirk Raatz and Oliver Reiser Tetrahedron 61 2005
Synthesis of the Novel Mannosidase Inhibitors (3R)- and (3S)-3-(Hydroxymethyl) swainsonine Erik J. Hembre and Willimn H. Pearson Tetrahedron 53 1997
A New Approach to Indolizidine Alkaloids: Asymmetric Formal Total Synthesis of (-) -Swainsonine Wei-Shan Zhou, Wen-Ge Xie, Zhi-Hui Lu, Xin-Fu Pan Tetrahedron Letters 36, No. 8 1995
Alpha-Acylamino Radical Cyclizations: Application to the Synthesis of (-) - Swainsonine Jeffrey M. Dener, David J. Hart, and Subban Ramesh J. Org. Chem. 53, No. 26 1988
New Chiral Route to (-)-Swainsonine via an Aqueous Acylnitroso Cycloaddition Approach Masaichi Naruse, Sakae Aoyagi, and Chihiro Kibayashi J. Org. Chem. 59 1994
Diastereoselective synthesis of new polyhydroxylated indolizidines from (L)- glutamic acid Katarina Kadlecikova, Vincent Dalla, Stefan Marchalin, Bernard Decroix and Peter Baranc Tetrahedron 61 2005
Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis. Alexander Deiters and Stephen F. Martin Chem Rev. 104(5) 2004
An Enantiocontrolled Synthesis Of (-)-Swainsonine Sung Ho Kang and Geun Tae Kim Tetrahedron Letters 36, No. 28 1995
Synthesis and mannosidase inhibitory activity of 3-benzyloxymethyl analogs of swainsonine William H. Pearson and Luyi Guob Tetrahedron Letters 42 2001
Stereochemical Issues ¶
Why chiral drugs ¶.
Biological receptors are chiral and involve a high degree of stereoselectivity. Chiral molecules are often used in drug research because of their 3D molecular arrangement, which is particularly adapted for meeting the stereochemical requirements of the receptor.

Examples of Chiral Drugs ¶
Many drugs are chiral. In fact, over 50% of the top-selling 500 pharmaceutical drugs on the market in 2004 were chiral molecules. Examples of some well known chiral drugs are shown below.

Multiple Aspects of Molecular Chirality ¶
The stereochemical course of a reaction is influenced by factors such as reactant and transition state energies, as well as the conformational and electronic properties. These properties are extensively presented in other chapters of this course, and the reader is invited to review them for more detail. In the following pages we focus on synthetic methods for preparing chiral molecules.

Active and Inactive Enantiomers ¶
As a consequence of the difference in molecular shape of enantiomers and the high degree of chirality of biological receptors, only one enantiomer is generally the active principle.
'Chiral Switch' : From a Racemic to a Chiral Drug ¶
In the late 1980s, most chiral drugs were commercialized as racemic mixtures. In many cases, one of the enantiomers was found to have more favorable therapeutic and pharmacokinetic characteristics only after commercialization of the racemate . A new trend known as 'chiral switching' has recently emerged, which aims at the development of single enantiomers of commercial racemate drugs. Since 1990, the proportion of single- enantiomer drugs approved as new chemical entities has been consistently greater than that of racemates . Today, market approval of a racemate has become much more stringent.

The Chiral Switch: The Development Of Single Enantiomer Drugs From Racemates Hutt, A.J. and Valentova, J. Acta Facult. Pharm. Univ. Comenianae 50 2003
Advantages of Single Enantiomer Drugs ¶
The advantages of single enantiomer drugs are multiple. These involve its pharmacokinetic properties, selectivity and patent related aspects, all of which are detailed in the following pages.

Pharmacokinetic Properties ¶
Because of their different 3D configurations, the pharmacodynamic and pharmacokinetic properties of enantiomers are likely to be different. When this is found to be true and one of the enantiomers has greater therapeutic and pharmacokinetic characteristics, a single enantiomer drug presents health benefits including an improved safety margin and other advantages such as those indicated below.

Perhexiline ¶
Perhexiline (an anti-angina drug), is an example of the different pharmacokinetic properties of enantiomers . The racemic of perhexiline was withdrawn from the market because it killed individuals in whom the wrong enantiomer was more slowly metabolized and who accumulated gram quantities of it. If the developers of the racemate had been aware of this noxious effect, they would have marketed a drug with only the fast-clearing enantiomer .

Selectivity ¶
When one enantiomer is responsible for the activity of interest, its paired 'inactive' enantiomer also has properties and effects that may not be negligible. It can interact with other receptors, cause side effects and be toxic. Some examples to illustrate this are given in the following pages.

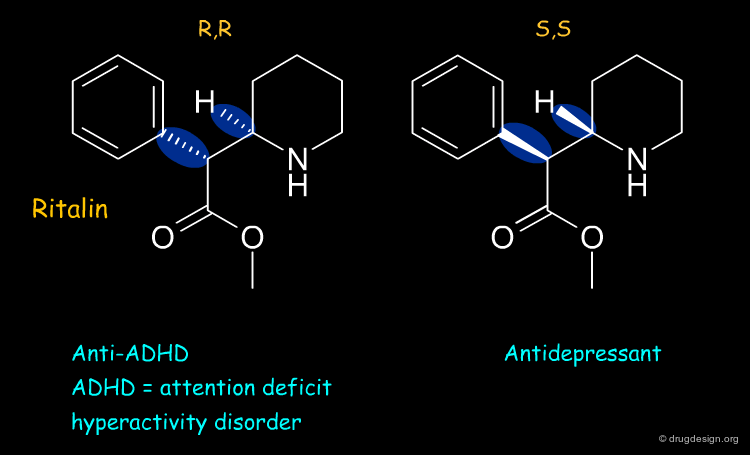
Ritalin ¶
Ritalin is used to treat attention deficit hyperactivity disorder (ADHD). The (R,R) enantiomer of ritalin has the anti-ADHD effect whereas the (S,S) form acts as an antidepressant.
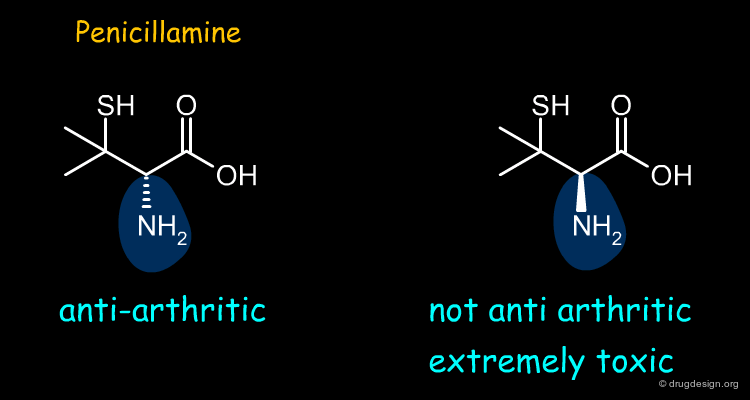
Penicillamine ¶
Penicillamine is an anti-arthritic drug. Most of the anti-arthritic activity of penicillamine resides in the (S)- enantiomer whereas the (R) form is extremely toxic.
Ethambutol ¶
Ethambutol is a tuberculostatic drug. The (S,S) enantiomer of ethambutol is a tuberculostatic agent whereas the (R,R) enantiomer is toxic (causes optical neuritis that can lead to blindness).
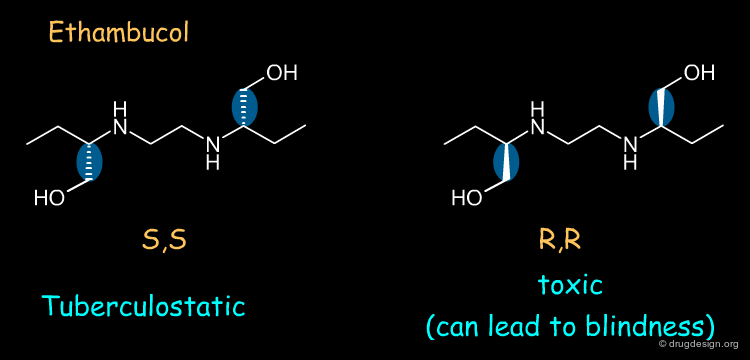
Ketamine ¶
Ketamine is an anesthetic drug. The S- enantiomer of ketamine possesses the anesthetic activity; the R- enantiomer can cause hallucinations.

L-Dopa ¶
Levodopa (L-dopa) is an anti-Parkinsonian drug that is commercialized in an enantiomerically pure form because the D-form is toxic and causes serious side effects (such as granulocytopenia).

Patent Position ¶
Chiral switching can be a useful marketing strategy for a company aiming at extending its patent franchise and interested in protecting itself from generic companies. For example the AstraZeneca patent for the ulcer drug Prisolec expired in 2002; the company successfully launched Nexium in 2001, the S- enantiomer of Prisolec. This enabled the company to keep its share of the market intact.

Three Methods to Obtain Chiral Molecules ¶
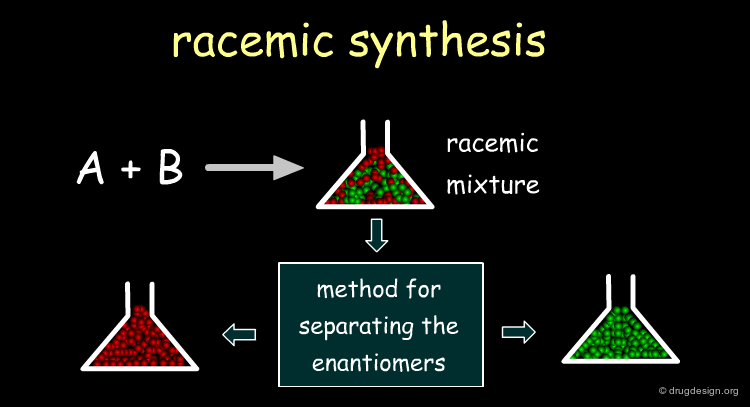
There are three approaches of obtaining a chiral molecule. The first consists of the synthesis of the racemate , followed by the separation of the enantiomers . The second is an asymmetric route for preparing the molecule with the proper stereochemistry. The third approach consists of extracting the chiral principle from natural sources (plants, animals).

Racemic Route ¶
It is usually simpler to develop a racemic synthesis than an asymmetrically pure one, and afterwards find a method for separating the enantiomers . In drug discovery it is crucial to rapidly obtain pure enantiomers for pharmacological tests, and the racemic route is preferred. Moreover, resolution methods enabling access to both enantiomers are also useful when preclinical studies are required for each enantiomer .
Asymmetric Catalysis in the Pharmaceutical Industry Joel M. Hawkins, Timothy J. N. Watson Angew. Chem. Int. Ed. Engl. 43, Issue 25 2004
Development of the Commercial Process for Zoloft/Sertaline George J. Quallich Chirality 17 2005
Resolution of Racemates ¶
In the mid 1980s enantiomers were separated by first forming salts or derivatives with chiral reagents, and then resolving them by conventional methods such as crystallization or chromatography on silica. Chromatography methodologies have now become the most widely utilized approach for the resolution of racemates . New developments such as countercurrent, supercritical fluid and simulated moving bed continuous chromatography have begun to revolutionize the drug development process in the industry.

Example of Industrial Racemate Separation ¶
Zoloft (compound 4), also known as sertraline is a chiral antidepressant drug marketed as a 1S(+),4S(+) enantiomer . Its synthesis is based on the key chiral tetralone 2. This building block is prepared industrially with high purity and yield from racemate 1, using simulated moving bed (SMD) continuous chromatographic technologies and racemization of the undesired enantiomer .
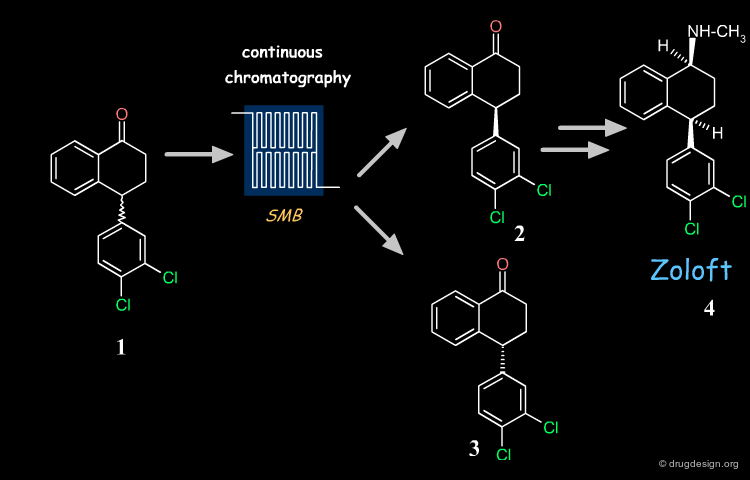
Advantages of the Process Chosen ¶
The industrial synthesis currently used for Zoloft eliminated the need for reagents such as titanium chloride and D-mandelic acid used in the initial inefficient route, and also removed the uncertainties surrounding royalties for particular reagents introduced by the medicinal chemists. It saved solvents, reagents and plant capacity previously harnessed for processing the undesired enantiomer , and resulted in immediate cash savings for Pfizer.

Recycling Undesired Enantiomer ¶
If the undesired enantiomer can be re-racemized, this enables all the racemate to be progressively converted into the desired isomer . There are many industrial examples of such recycling. One example is the tetralone intermediate involved in the synthesis of Zoloft whose unwanted enantiomer is repeatedly recycled, as visualized below. With this procedure the desired enantiomer is prepared industrially with high purity and yield ( ee = 99%; recovery yield 98%).

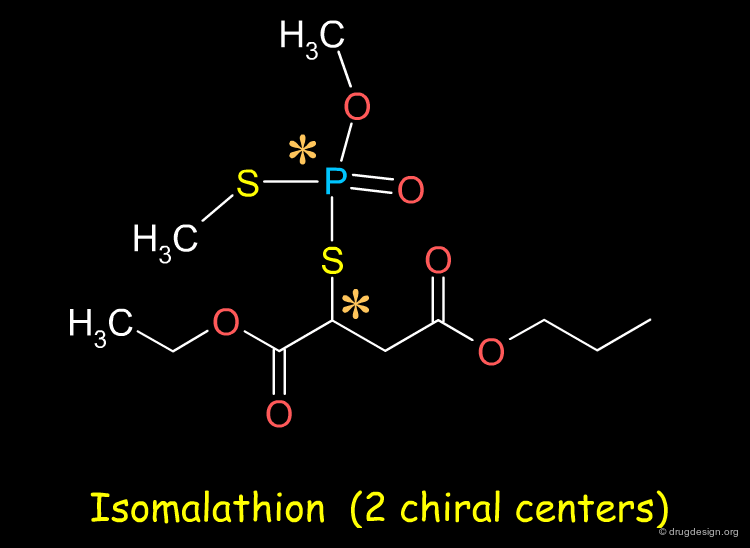
Sometimes Separation very Difficult ¶
The separation of enantiomers can be very challenging. For example the resolution of the four stereoisomers of the organophosphoramidate compound isomalathion is a difficult problem: for some compounds it may not be straightforward to separate the enantiomers .
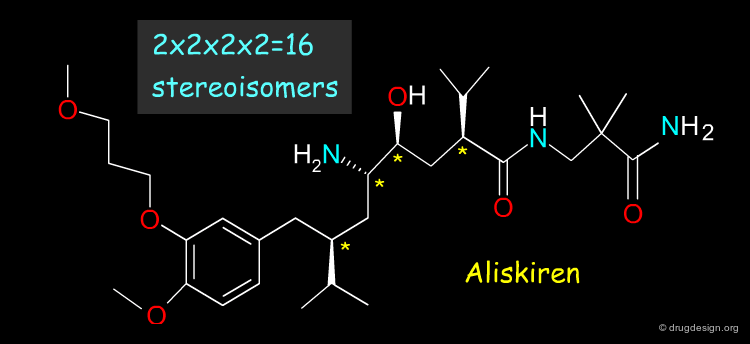
When Too Many Chiral Centers ¶
The racemic route is not realistic when the molecule contains too many chiral centers; for example Aliskiren (a renin inhibitor now in late stages of development) has 4 asymmetric centers. For molecules with many chiral centers a stereoselective route is necessary.
Asymmetric Route ¶
Asymmetric synthesis aims at the direct preparation of the target molecule with the proper chirality. Brilliant molecule builders such as Woodward, Corey, Kishi, Nicolaou and others succeeded in synthesizing complicated chiral molecules with intelligent science and fine art. Thanks to their heroic efforts, an expanding arsenal of tools is now available in synthetic chemistry for the preparation of chiral molecules. Chirality can be introduced either through chiral reagents or chiral catalysts.

Chiral Reagents and Reactants ¶
Aliskiren is shown here to illustrate the asymmetric synthesis of a drug. Lactone 2 is a key building block; its opening gives the proper stereochemistry for both the hydroxyl and the isopropyl groups located on the right side of the structure. Likewise the chirality of the second isopropyl and that of the amino are also introduced stereospecifically (not shown here). All four chiral centers of Aliskiren are created by asymmetric syntheses.

The Power of Visual Imagery in Synthesis Planning Stereocontrolled Approaches to CGP-60536B, a Potent Renin Inhibitor S. Hanessian, S. Claridge, and S. Johnstone J. Org. Chem. 67 2002
A Convergent Synthesis of the Renin Inhibitor SPP-100 Using a Nitrone Intermediate Alessandro Dondoni, Geert De Lathauwer and Daniela Perrone Tetrahedron Letters 42 2001
A Convergent Synthesis of the Renin Inhibitor CGP60536B David A. Sandham, Roger J. Taylor, John S. Carey and Alexander Faessler Tetrahedron Letters 41 2000
A Convergent Synthesis Approach Towards CGP60536B, a Non-Peptide Orally Potent Renin Inhibitor, Via An Enantiomerically Pure Ketolactone Intermediate Heinrich Rueeger, Stefan Stutz, Richard Goeschke, Felix Spindler and Juergen Maibaum Tetrahedron Letters 41 2000
Novel 2,7 Dialkyl Substituted 5(S)-Amino-4(S)-Hydroxy-8-Phenyl-Octanecarboxamide Transition State Peptidomimetics are Potent and Orally Active Inhibitors of Human Renin R. Goeschke, V. Rasetti, N.C. Cohen, J. Rahuel, M. Gruetter, S. Stutz, H. Rueger, J.M. Wood, J. Maibaum XVth Meeting of the European Federation of Medicinal Chemistry, Edinburg
Design and Syntheses of Novel 2,7Dialkyl substituted 5(S)Amino 4(S)Hydroxy-8Phenyl- Octanecarboxamides-as in vitro Potent Peptidomimetic Inhibitors of Humain Renin R. Goeschke, N.C. Cohen, J.M. Wood and J. Maibaum Bioorganic and Med. Chem. Letters 7 1997
Chiral Catalysts ¶
Chiral catalysts such as metal complexes or enzymes can be used to create new asymmetric centers starting from prochiral molecules. In the example below the painkiller S-Naproxen is obtained with good yield and purity using Ru-S-BINAP as a catalyst for the hydrogenation.

Asymmetric Synthesis in R&D. ¶
The development of an asymmetric synthesis is generally not considered in the early drug discovery stage, since it is both time-consuming and expensive: whenever possible the racemic route is preferred. For large scale production of pure stereo- isomers , the set up and optimization of an asymmetric process still requires substantial experimentation and expensive catalysts, with the important constraint of being economically acceptable.

Extraction From Natural Sources ¶
Besides the therapeutic advantages of single enantiomers , their industrial manufacturing is an important issue. For example pain relieving morphine has five asymmetric centers. The molecule is synthesized by the poppy plant Papaver somniferum. On the other hand Woodward and other groups successfully achieved the total synthesis of this alkaloid. However, the costs associated with the preparation of large amounts are such that the extraction approach is by far the more economical.
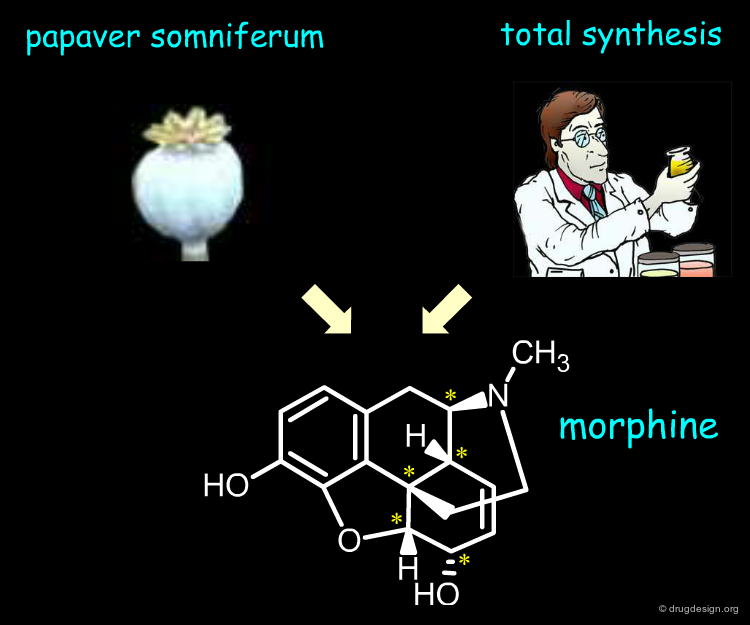
Synthesis of (-)-Morphine Douglass F. Taber, Timothy D. Neubert, and Arnold L. Rheingold J. Am. Chem. Soc. 124 2002
The Synthesis of Morphine M. Gates, G. Tschudi J. Am. Chem. Soc. 74 1952
The Synthesis of Morphine M. Gates, G. Tschudi J. Am. Chem. Soc. 78 1956
The Synthesis of Morphine D. Elad, D. Ginsburg J. Am. Chem. Soc. 76 1954
Biogenetically patterned synthesis of the morphine alkaloids M. A. Schwartz, I. S. Mami J. Am.Chem. Soc. 97 1975
Synthetic opium alkaloids and derivatives. A short total synthesis of (+-)- dihydrothebainone, (+-)-dihydrocodeinone, and (+-)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners K. C. Rice J. Org. Chem. 45 1980
Studies Directed Towards The Total Synthesis Of Morphine Alkaloids D. A. Evans, C. H. Mitch Tetrahedron Lett. 23 1982
Synthesis via vinyl sulfones. 21. Total synthesis of dl-morphine J. E. Toth, P. L. Fuchs J. Org. Chem. 52 1987
Convergent synthesis of (+-)-dihydroisocodeine in 11 steps by the tandem radical cyclization strategy. A formal total synthesis of (+-)-morphine K. A. Parker, D. Fokas J. Am. Chem. Soc. 114 1992
Asymmetric synthesis of either enantiomer of opium alkaloids and morphinans. Total synthesis of (-)- and (+)-dihydrocodeinone and (-)- and (+)-morphine C. Y. Hong, N. Kado, L. E. Overman J. Am. Chem. Soc. 115 1993
Formal Total Synthesis of ( - )-Morphine by Cuprate Conjugate Addition J. Mulzer, G. Duerner, D. Trauner Angew. Chem. Int. Ed. Engl. 35 1996
Stereoselectivity of Action not Always Predictable ¶
Although there are theoretical advantages in developing single enantiomer drugs, there are cases where the approach has failed. For example Dilevalol was thought to have advantages over the corresponding racemic drug Labetalol (a beta-blocker launched in 1977), but it was taken off the market because of toxicity. ADME /Tox is a science that is far from being fully understood; the pharmacokinetic and pharmacodynamic data of racemic mixtures and enantiomers can be ambiguous and mask unpredictable effects.

Spontaneous Enantiomer Interconversion ¶
Normally a stereospecific synthesis aims at obtaining the desired active enantiomer . Thalidomide is a special case where such effort was worthless. The drug was introduced in the 1950s as a sedative and was used by pregnant women to treat morning sickness. Birth defects were observed and Thalidomide was banned worldwide. Only the R enantiomer has the sedative action, whereas the S enantiomer was found to be teratogenic. However the active enantiomer racemizes in less than 10 minutes in the body and the tragedies could not have been avoided by use of the active R-form.

Structural Diversity and Higher Level Synthetic Strategies ¶
Diverse strategy in drug discovery ¶.
The space of synthesizable molecules is huge. Structural diversity is essential for lead discovery, and the generation of a great number of diverse molecules increases the chances of finding novel compounds with significant therapeutic value.

Introducing Diversity in the Reagents ¶
Diversity is always introduced by the reagents. In the example below, diverse compounds such as 1 need to be prepared. They are synthesized by the reaction of building block 2 with a number of diverse reagents that can be either purchased or simply prepared.

Introducing Diversity at the Proper Time ¶
The introduction of diversity through reagents should be made at the right time. Important considerations include when protection/deprotection of functional groups should take place and fully exploit the strength of the synthetic route, whether it is linear or convergent.
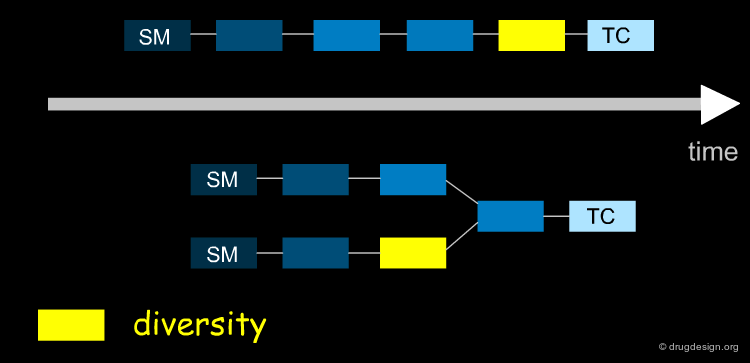
Late Stage Introduction of Diversity ¶
When variations are introduced at the end of the synthesis, many diverse analogs can be prepared in a minimum number of steps. This is the case of Aliskiren that was synthesized using the retrosynthetic scheme shown here (button "Retrosynthesis"). Hundreds of amide analogs such as 6 were prepared from acid 2 in order to optimize the series.

Early Stage Introduction of Diversity ¶
When variations are needed far from the end of the synthesis, diverse analogs are very difficult to prepare. This is the case of the aryl moiety of Aliskiren. Many reagents of structure 7 must be prepared and moreover, additional reactions are necessary before arriving at the target compounds 8. With this synthesis the preparation of many diverse aryl derivatives is extremely difficult.
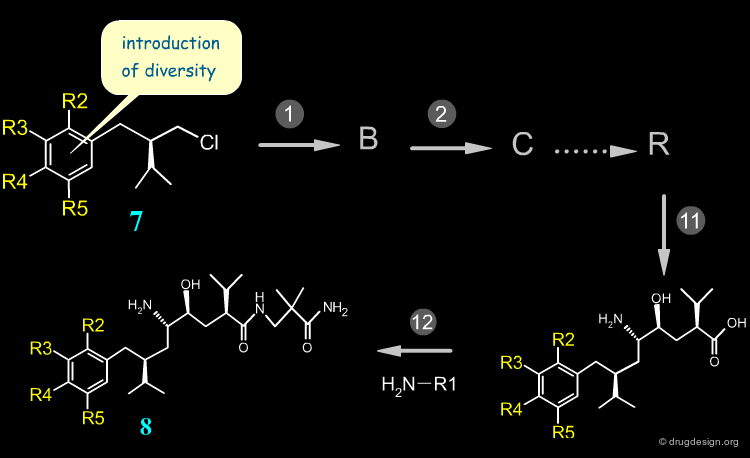
Diversity Space Restricted by Synthetic Route ¶
As chemistry is based on the transformation of functional groups, inevitably a given synthetic scheme restricts the field of the possible diverse analogs that can be synthesized. In theory, several reaction routes are needed to better cover the entire diversity space, but in practice this is not always feasible. This is discussed in the following pages.
Consider Alternative Syntheses ¶
One way of escaping the bias of a single synthetic scheme is to consider multiple routes. This may be important because a single change in a fragment may transform a micromolar hit into a nanomolar one. Very often the optimization of a lead is greatly restricted, the molecules prepared are not those that should be made and the optimization process is trapped with no substantial change in biological activities. Another synthesis is likely to open up new possibilities and reveal an entirely different moiety that can lead to potency and selectivity.

Example of Introduction of Diversity ¶
With the aim of incorporating diversity in a structure, it may be necessary to modify the initial lead. In the example below the Abbott group replaced the indane moiety of a SmithKline lead by a pyrrolidine. This novel structure permitted the synthesis of many analogs and the rapid generation of useful structure-activity relationships .

2,4-Diarylpyrrolidine-3-carboxylic acids--potent ETA selective endothelin receptor antagonists. 1. Discovery of A-127722. Winn M, von Geldern TW, Opgenorth TJ, Jae HS, Tasker AS, Boyd SA, Kester JA, Mantei RA, Bal R, Sorensen BK, Wu-Wong JR, Chiou WJ, Dixon DB, Novosad EI, Hernandez L, Marsh KC. J Med Chem. 39(5) 1996
Combinatorial Chemistry ¶
Combinatorial chemistry enables the creation of a large population of molecules (called libraries) that can be screened at one time. Typically, analogs of a common scaffold are synthesized in an automated manner (robotics) with a common synthetic route. Examples of libraries that can be created with these technologies are visualized below. By producing large and diverse compound libraries, the chemist increases the probability of finding novel compounds of interesting therapeutic value. The field of combinatorial chemistry is presented in detail in a special chapter in this course.

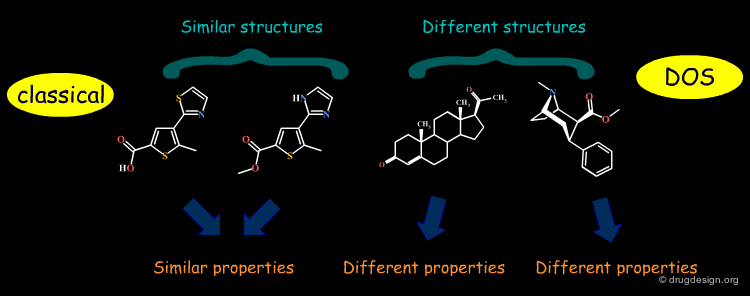
Diversity-Oriented Synthesis (DOS) ¶
The synthetic strategy commonly used in combinatorial chemistry is to access diverse collections of small molecules by appending different sets of building blocks ("appendage decoration") to a common molecular skeleton. The impossibility of considering the scaffold as a "variable" has triggered the development of a new approach in synthetic chemistry known as "diversity-oriented synthesis" (DOS) which also aims at varying scaffolds.

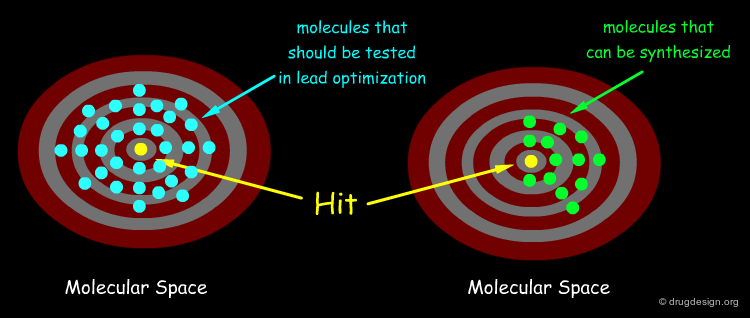
Maximizing the Chance to Find a Hit ¶
The quest for molecular diversity is based on the "similar property principle" which assumes that structurally similar molecules are likely to have similar properties and structurally dissimilar molecules are likely to have different properties. By enabling the variation of both scaffolds and substituents, DOS maximizes the chance of finding relevant hits.
Example of Diversity-Oriented Synthesis ¶
An example of a DOS reaction is given below that shows how an immobilized dihydroisoquinoline reactive intermediate is used to obtain skeletally diverse compounds.

Synthetic strategy toward skeletal diversity via solid-supported, otherwise unstable reactive intermediates. Taylor SJ, Taylor AM, Schreiber SL Angew Chem Int Ed Engl 43 2004
Higher Level Strategies in Organic Synthesis ¶
The creation of more than one target molecule in a diversity-driven approach was introduced by Stuart Schreiber at Harvard University. Since the means available in synthetic chemistry are not suited to this approach, it has prompted many research groups to attempt to create new types of synthetic planning methods. The sketch shown below of the areas now covered by organic synthesis, suggests that the search for higher level strategies will be an important driving force for the next generation of methodology-oriented synthetic organic chemists.

Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery Stuart L. Schreiber SCIENCE 287 17 March 2000
Generating Diverse Skeletons of Small Molecules Combinatorially Martin D. Burke, Eric M. Berger, Stuart L. Schreiber SCIENCE 302 24 October 2003
The small-molecule approach to biology Stuart L. Schreiber Chem Eng News 81 2003
A synthesis strategy yielding skeletally diverse small molecules combinatorially. Burke MD , Berger EM, Schreiber J Am Chem Soc 126 2004
A planning strategy for diversityoriented synthesis. Burke MD , Schreiber SL Angew Chem Int Ed Engl 43 2004
Diversity-Oriented synthesis of biaryl-containing medium rings using a one bead/one stock solution platform. Spring DR, Krishnan S, Blackwell HE, Schreiber SL J Am Chem Soc 124 2002
Synthesis of Some Common Drugs ¶
Introduction to synthetic schemes ¶.
In the previous pages the main principles that govern the synthesis of novel drugs were discussed. In this section real examples are presented and include the following information: the chemical formula of the drug, a retrosynthetic analysis, the synthesis and some physico-chemical properties of the drug. We start with benzocaine.

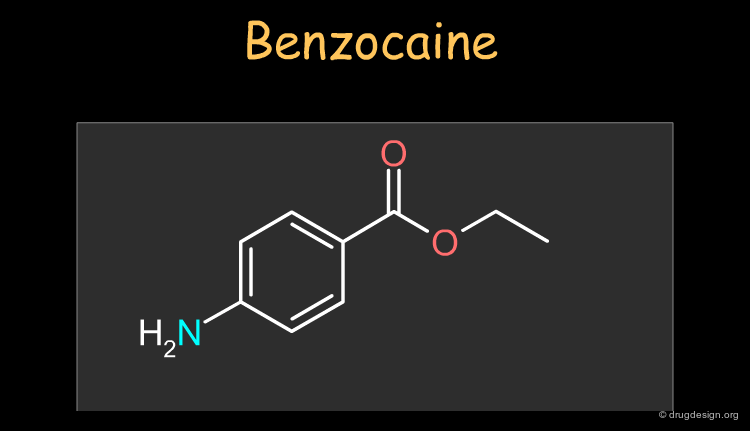
Benzocaine ¶
Benzocaine is a widely used local and topical anesthetic. The drug can be synthesized as described in the following pages.
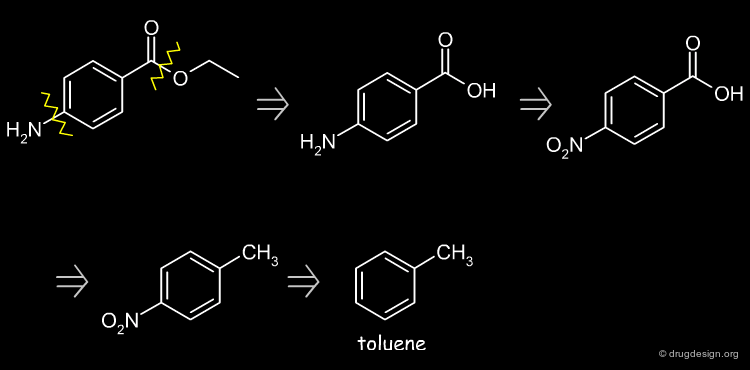
Retrosynthetic Scheme ¶
Possible starting disconnections are the ester or the amine groups. The amine disconnection leads to toluene as starting material.
Benzocaine Synthesis ¶
Nitration of toluene, oxidation of the methyl group, reduction of the nitro and subsequent esterification of the carboxylic acid function leads to the desired product.

Clayden, Greeves, Warren and Wothers Organic Chemistry Oxford University Press 2001
Physico-Chemical Properties of Benzocaine ¶
Some physico-chemical properties of benzocaine are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

Aspirin ¶
Aspirin is a very popular analgesic agent with a relatively simple structure. An outline of the story associated with the discovery of aspirin can be found in a special chapter in this course. This drug can be synthesized as described in the following pages.

Aspirin Synthesis ¶
Salicylic acid can be the starting material for aspirin since it is a readily available compound. A simple esterification step leads to the desired drug.

K.L. Williamson Macroscale and Microscale Organic Experiments D.C. Health and Company. 1989
Physico-Chemical Properties of Aspirin ¶
Some physico-chemical properties of aspirin are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

Nalidixic Acid ¶
Nalidixic acid is a synthetic chemotherapeutic agent which works against gram-negative bacteria. The drug can be synthesized as described in the following pages.

A possible retrosynthetic scheme for the synthesis of nalidixic acid is presented below; it is based on a substituted pyridine as starting material.

Nalidixic Acid Synthesis ¶
Nalidixic acid can be synthesized in three steps from methyl-2, amino-6 pyridine, as described in the synthetic route below.
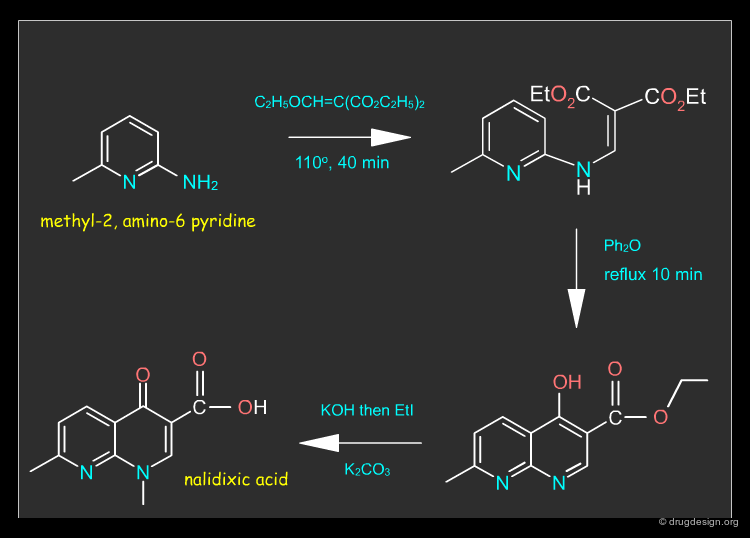
8-Naphthyridine Derivatives. A New Class of Chemotherapeutic Agents George Y. Lesher, Ernest J. Froelich, Monte D. Gruett, John Hays Bailey, and R. Pauline Brundage J. Med. Chem. 5 1962
Physico-Chemical Properties of Nalidixic Acid ¶
Some physico-chemical properties of nalidixic acid appear below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

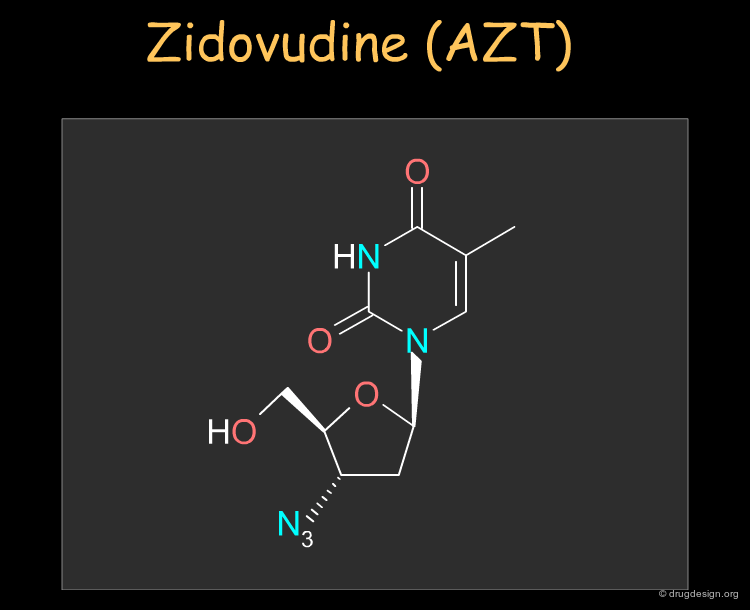
Zidovudine (AZT) ¶
Zidovudine (AZT) was the first nucleoside reverse transcriptase inhibitor antiviral agent discovered and used to treat human immunodeficiency virus (HIV) that causes AIDS. The drug can be synthesized as described in the following pages.
The retrosynthetic scheme shown below indicates that AZT 1 can be synthesized in four steps, starting from building block 5.

AZT Synthesis ¶
In the first step the primary alcohol of (2'-deoxy-lyxo furanosyl) thymine 1 is protected selectively with trityl chloride. In the second step the secondary alcohol is activated, using MeSO2Cl and a nucleophilic displacement reaction. Treatment of the mesyl derivative with lithium azide leads to product 4. The trityl group is then deprotected under acidic conditions to give AZT.

Nucleosides. V. The Monomesylates of 1-(2?-Deoxy-?-D-lyxofuranosyl)thymine Jerome P. Horwitz, Jonathan Chua, and Michael Noel J. Org. Chem. 29 1964
Nucleotide synthesis. IV. Phosphorylated 3'-amino-3'-deoxythymidine and 5'-amino-5'-deoxythymidine and derivatives Ronald P. Glinski, M. Sami Khan, Richard L. Kalamas, and Michael B. Sporn J. Org. Chem. 38(25) 1973
** ** C.K. Chu et al. Tetrahedron Letters 29 1988
Another Synthesis for AZT ¶
AZT can be synthesized in different ways. Another example of synthesis is given below, which starts from D-mannitol. The key reactions in this scheme are Witting and Michael reactions. Move the cursor over the 'i' symbols to display the reaction conditions.

** ** C.K. Chu et al Tetr. Lett. 29 1988
Which Route is Preferable? ¶
The synthesis that starts from D-mannitol is more economic. Thymidine the starting material for the first scheme is very expensive whereas D-mannitol is readily available. Today not only economic factors but also ecologic factors are taken into account and chemists are paying more attention to green chemistry.

Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice Oxford University Press, New York 1998
Physico-Chemical Properties of Zidovudine ¶
Some physico-chemical properties of Zidovudine are presented below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

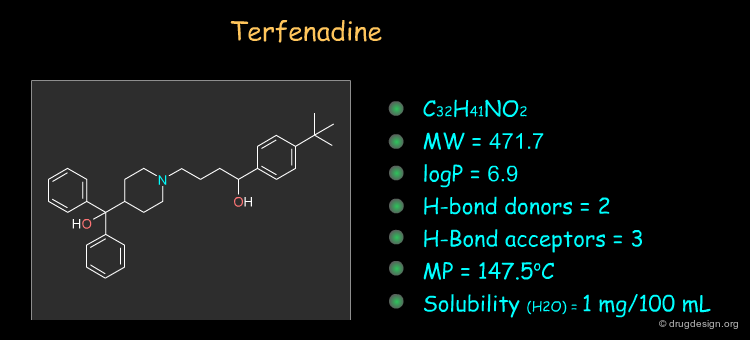
Terfenadine ¶
Histamine is released in a variety of allergic conditions and mediates its activity by interaction with H1 receptors. Terfanidine is H1 blocker of the second generation and is used for the treatment of seasonal allergic rhinitis. The drug can be synthesized as described in the following pages.

A possible retrosynthetic scheme for the synthesis of Terfenadine is indicated below. The synthesis is based on the substituted piperidine 3, and the chloro-ketone 4.
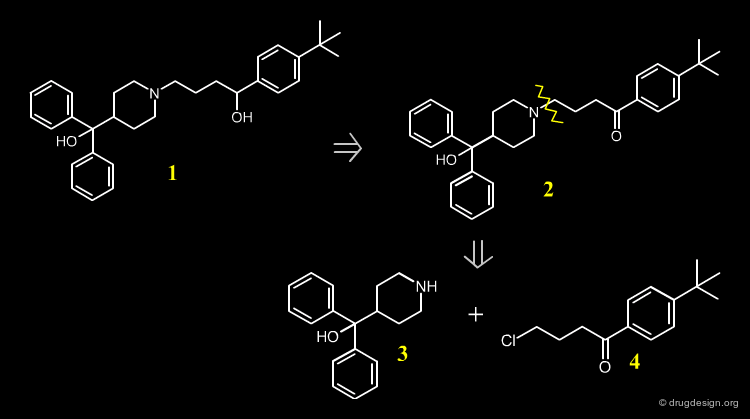
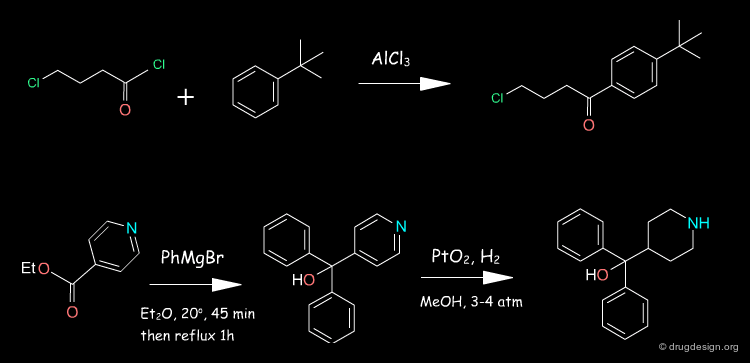
Terfenadine Synthesis ¶
The 4-pyridine methanol is obtained using isonicotinic ethyl ester and a double Grignard reaction with phenyl magnesium bromide. Hydrogenation follows, in the presence of PtO2. The obtained azacyclonol is alkylated with the chlorobutanone in refluxing toluene in the presence of an inorganic base and a catalytic amount of NaI. Terfenadine is obtained using a reducing agent KBH4 as a racemic alcohol.
Terfenadine Albert A. Carr et al Arzneim Forsch 32 1982
Physico-Chemical Properties of Terfenadine ¶
Some physico-chemical properties of terfenadine are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.
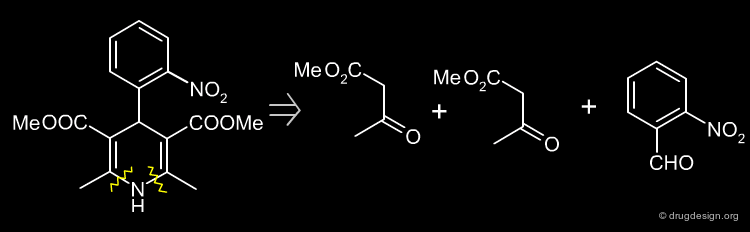
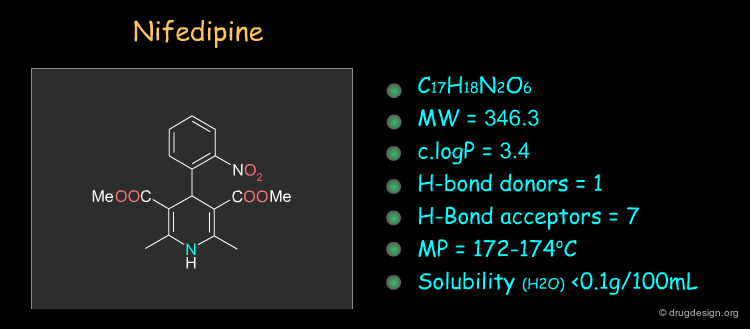
Nifedipine ¶
Nifedipine was the first drug approved in the dihydropyridine series. It is a calcium channel blocker and it acts in the treatment of angina and hypertension. This drug can be synthesized as described in the following pages.

Simple Retrosynthesis of Nifedipine ¶
The following scheme describes a simple retrosynthesis of Nifedipine starting from 2-nitrobenzaldehyde.
One-Pot Synthesis of Nifedipine ¶
The corresponding synthesis of Nifedipine can be developed in a 'one-pot' reaction (Hantzsch condensation). However, it yields several products and therefore is not optimal.

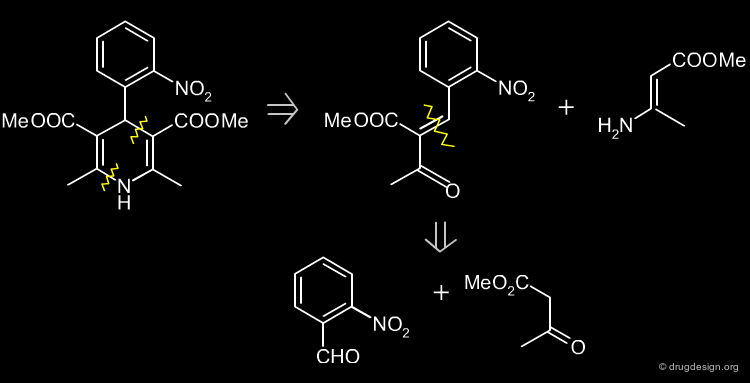
Two Steps Retrosynthesis Scheme of Nifedipine ¶
The retrosynthesis of Nifedipine presented here involves two steps, but is more preferable because it does not have the drawback of impurities.
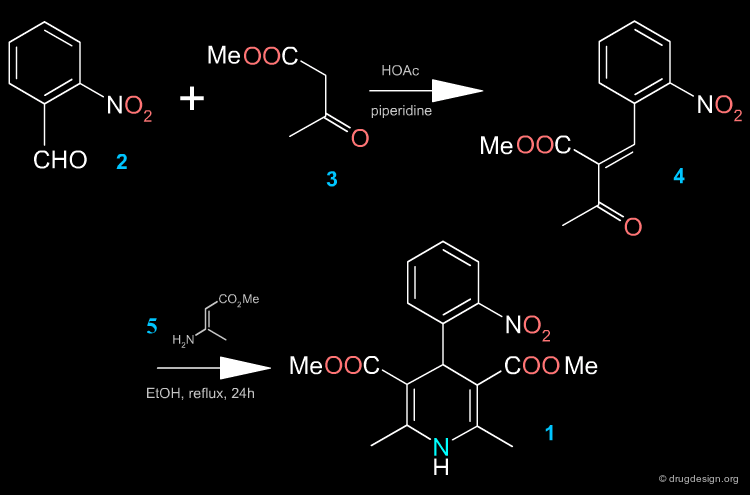
Two Steps Synthesis of Nifedipine ¶
In the first step, the benzylidine ester 4 is produced (intermediate Knoevenagel product). A Hantsch reaction follows with the ethyl 3-aminocrotonate 5, which leads to the desired product.
Physico-Chemical Properties of Nifedipine ¶
Some physico-chemical properties of Nifedipine are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.
Sildenafil (Viagra) ¶
Sildenafil (Viagra) is a drug used to treat impotence in men. The molecule can be synthesized as described in the following pages.

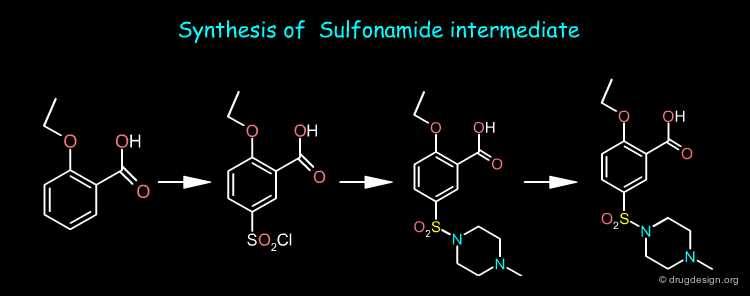
A retrosynthesis of Sildenafil involves only two precursors: the nitropyrazole and the sulfonamide intermediates shown below.

Sildenafil Synthesis ¶
The nitropyrazole intermediate can be prepared from methylhydrazine, estermalonate and 2-pentanone as starting materials whereas the sulfonamide intermediate is prepared from o-ethoxy benzoic acid. Sildenafil is obtained by first reducing the nitro of the nitropyrazole and then coupling the two pieces and cyclization of the pyrimidone ring.
The development of an environmentally benign synthesis of sildenafil citrate (Viagra ) and its assessment by Green Chemistry metrics Peter J. Dunn, Stephen Galvin and Kevin Hettenbach Green Chemistry 6(1) 2004
Physico-Chemical Properties of Sildenafil ¶
Some physico-chemical properties of Sildenafil are listed below. The molecular weight, logP, the number of H-bond donors and H-bond acceptors can be compared to Lipinski's rule of five: all the values are within the framework of this rule.

Programs for Computer-Aided Synthesis ¶
Programs for computer-aided synthesis started to appear in the late 1960s. Important progress has been made; however system development was hampered by the reluctance of chemists to use such programs. Useful for the exploration of synthetic routes, these programs may be disappointing when utilized as black boxes to find a relevant route for the synthesis of a complex molecule. A brief outline of some of these systems is given in the following pages.

Computer-assisted Synthesis N.S. Zefirov and E.V. Gordeeva Uspekhi Khimii 56(10) 1987
Cheminformatics and Organic Chemistry. Computer-Assisted Synthetic Analysis Martin Ott Cheminformatics (IOS Press) 1 Numbers 1-2 2004
Computer-aided organic synthesis Mattwew Todd Chem Soc Rev 34(3) 2005
R. Barone, M. Chanon Encyclopedia of Computational Chemistry John Wiley and Sons, Chichester 1998
Retrosynthetic Programs ¶
When a chemical reaction is viewed "retrosynthetically" it is considered as going backwards from a target molecule to the starting materials. The main aim of retrosynthetic strategies is to generate precursors that correspond to available starting material. In the following pages examples of retrosynthetic programs are presented.

LHASA ¶
LHASA (Logic and Heuristics Applied to Synthetic Analysis) was developed in the early 1960s in a group headed by E.J. Corey at Harvard, and was one of the first major systems made for computer-assisted synthetic design. It was initially developed to implement Corey's rules for synthetic chemistry (1967) where he defined novel concepts such as 'retrosynthesis', 'synthons' and 'disconnections'. Since its introduction LHASA has been actively maintained and enhanced. The system was and continues to be used by many groups worldwide.

Algorithm for machine perception of synthetically significant rings in complex cyclic organic structures E. J. Corey and George A. Petersson J.Am.Chem.Soc. 94 1972
General methods of synthetic analysis. Strategic bond disconnections for bridged polycyclic structures E. J. Corey, W. Jeffrey Howe, H. W. Orf, David A. Pensak, and George Petersson J.Am.Chem.Soc. 97 1975
Computer-assisted synthetic analysis. Synthetic strategies based on appendages and the use of reconnective transforms E. J. Corey and William L. Jorgensen J.Am.Chem.Soc. 98 1976
SYNCHEM ¶
SYNCHEM was developed in the early 1970s by H.L. Gelernter at Sunny Brook (NY) for automatic organic synthesis generation using artificial intelligence and heuristic computer programming. The method was based on a knowledge base containing data that were published on the chemical reactions involved and a small catalogue of readily available starting material. Challenged by synthetic experts the program was criticized for giving "naive" solutions too often. Attempts were made in 1978 to improve the first version.

Empirical Explorations of SYNCHEM Gelernter, H.L., Sanders, A.F., Larsen, D.L., Agarwal, K.K, Boivie, R.H., Spritzer, G.A., and Searleman, J.T. Science 197 Sept. 9, 1977
A Computer-Oriented Linear Canonical Notational System for the Representation of Organic Structures with Stereochemistry. Krishna K. Agarwal, Herbert L. Gelernter Journal of Chemical Information and Computer Sciences 34(3) 1994
Building and refining a knowledge base for synthetic organic chemistry via the methodology of inductive and deductive machine learning Herbert L. Gelernter, J. Royce Rose, Chyouhwa Chen Journal of Chemical Information and Computer Sciences 30(4) 1990
Application of Chemical Transforms in SYNCHEM2 K. Agarwal, D. Larsen, and H. Gelernter Computers and Chemistry 2 1978
A Computer-Oriented Linear Canonical Notational System for the Representation of Organic Structures with Stereochemistry K. Agarwal and H. Gelernter Journ. Chem. Inf. Comp. Sci. 34 1994
SECS, OCCS, CASP ¶
SECS (Simulation and Evaluation of Chemical Synthesis) was developed in 1978 by W.T. Wipke. The program recognizes stereochemical features. In addition 3D energy minimizations can be performed to assess whether a reactive site is accessible or subject to a particular steric hindrance before a suitable transform is proposed. The predecessor of SECS was OCCS (Organic Chemical Simulation of Synthesis - 1969). The CASP (Computer Aided Synthesis Planning System) system developed and used in the 1980s by a consortium of European pharmaceutical companies is an offshoot of the SECS program.

Simulation and Evaluation of Chemical Synthesis - SECS: An Application of Artificial Intelligence Techniques W. Todd Wipke, Glenn I. Ouchi, S. Krishnan Artif. Intell. 11(1-2) 1978
Formal and Mathematically-Based Programs ¶
Formal and mathematically based programs use theoretical descriptions of possible chemical reactions. The formalism can be based on matrices, a set of heuristics or symbolic representations of bond formation and cleavage. In the following pages some programs based on such formal descriptions of chemical reactions are described.

IGOR ¶
IGOR (Interactive Generation of Organic Reactions) was developed in the late 1970s by I. Ugi as a logic-based synthetic analysis program. A molecule is represented as a matrix; chemical reactions are also represented by matrices. The product of a reaction is obtained by adding the corresponding reaction matrix to the starting material matrix. The mathematical framework provides a convenient way to describe and classify reactions. IGOR contributed to the discovery of new reactions, which were later shown to be possible experimentally. The program is interactive and driven by the user who provides chemical knowledge, intuition and preferences.

Computer-Assisted Solution of Chemical Problems: A New Discipline in Chemistry Ivar Ugi, Johannes Bauer, Klemens Bley, Alf Dengler, Andreas Dietz, Eric Fontain, Bernhard Gruber, Rainer Herges, Michael Knauer, Klaus Reitsam, Natalie Stein Angew. Chem., Int. Ed. Engl. 32 1993
EROS ¶
EROS (Elaboration of Reactions for Organic Synthesis) was developed in the late 1970s by the group of J. Gasteiger in Germany. It was designed as a reaction generation program with the intention of limiting the combinatorial explosion of the synthetic tree. The acceptance or the rejection of reaction steps is based on thermodynamic considerations (reaction enthalpies, bond dissociation energies) and electronic parameters (atomic charges, inductive, resonance, polarizability effects). EROS contributed to the discovery of a simple industrial route to Acrylonitrile.

EROS - A Computer Program for Generating Sequences of Reactions J. Gasteiger, C. Jochum Topics Curr. Chem. 74 1978
Computer-assisted Reaction Prediction and Synthesis Design J. Gasteiger, W.-D. Ihlenfeldt, P. Roese, R. Wanke Anal. Chim. Acta 235 1990
A New Treatment of Chemical Reactivity: Development of EROS, an Expert System for Reaction Prediction and Synthesis Design J. Gasteiger, M. G. Hutchings, B. Christoph, L. Gann, C. Hiller, P. Loew, M. Marsili, H. Saller, K. Yuki Topics Curr. Chem. 137 1987
SYNGEN ¶
SYNGEN (SYNthesis GENeration) is a program introduced in the late 1970s by J.B. Hendrickson at Brandeis where he developed a novel approach for the generalization of organic functionality and reactions based on a symbolic representation of bond formation and cleavage. The program tries to find all the most efficient routes - the 'ideal synthesis' being the shortest and most atom-economical. SYNGEN is accompanied by another program enabling the interactive analysis of synthetic routes.

Organic Synthesis in the Age of Computers James B. Hendrickson Angew. Chem., Int. Ed. Engl. 29, Issue 11 November 1990
A general protocol for systematic synthesis design James B. Hendrickson Top Curr Chem. 62 1976
A logic-based program for synthesis design James B. Hendrickson, David L. Grier, and A. Glenn Toczko J.Am.Chem.Soc. 107 1985
Comprehensive System for Classification and Nomenclature of Organic Reactions James B. Hendrickson J.Chem.Inf.Comput.Sci. 37 1997
RAIN ¶
RAIN (Reactions And Intermediates Networks) is a reaction generator program that generates reactions and reaction schemes from reactions and structures by taking both the synthetic and retrosynthetic directions into account. RAIN was introduced by the Ugi group in 1987 using the same algebraic model of constitutional chemistry as in IGOR. The method is useful for elucidating reaction mechanisms.

Computer Assisted Bilateral Generation of Reaction Networks from Educts and Products E. Fontain, J. Bauer, I. Ugi Chem. Lett.
The Generation of Reaction Networks with RAIN. 1. The Reaction Generator E. Fontain, K. Reitsam J. Chem. Inf. Comput. Sci. 31 1991
The Generation of Reaction Networks with RAIN. 2. Resonance Structures and Tautomerism E. Fontain Tetrahedron Comput. Methodol. 3 1990
Forward Reaction Predictions ¶
Forward reaction predictions correspond to approaches that develop in the synthetic direction; therefore they begin with the starting material. Examples of starting-material oriented search programs are presented in the following pages.

CAMEO ¶
CAMEO (Computer-Assisted Mechanistic Evaluation of Organic Reactions) is a program developed in the early 1980s by W.I. Jorgensen for the prediction of the mechanism and products of an organic reaction. The program is instructed for the identification of electrophiles, nucleophiles and pKa values. Given the starting materials, reagents and conditions, the goal of the program is to predict all possible products that may be produced. CAMEO has been tested successfully against a number of published synthetic works.

CAMEO: a program for the logical prediction of the products of organic reactions William L. Jorgensen, Ellen R. Laird, Alan J. Gushurst, Jan M. Fleischer, Scott A. Gothe, Harold E. Helson, Genevieve D. Paderes, and Shenna Sinclair Pure and Applied Chem. 62 (10) 1990
Computer-assisted mechanistic evaluation of organic reactions. 1. Overview Timothy D. Salatin and William L. Jorgensen J.Am.Chem.Soc. 45 1980
CHIRON ¶
CHIRON was developed by S. Hanessian (1990) as a starting material-oriented program to reveal appropriate starting material. The program searches for maximal similarity in terms of carbon skeleton. Stereochemistry and functionality, is searched by the program between the target and databases of available chemicals. CHIRON has proven capable of finding starting material for complex targets with many chiral centers.

The Psychobiological Basis of Heuristic Synthesis Planning -Man, Machine and the Chiron Approach S. Hanessian, J. Franco and B. Larouche Pure and Applied Chem. 62 1990
Computer-Assisted Analysis and Perception of Stereochemical Features in Organic Molecules Using the Chiron Program S. Hanessian, J. Franco, G. Gagnon, D. Larame and B. Larouche J. Comp. Chem. Inf. Sci. 30 1990
WODCA ¶
WODCA (Workbench for the Organization of Data for Chemical Applications) was developed in 1995 by J. Gasteiger's group in Germany. This software incorporated the EROS reaction generation program into the broader WODCA range of programs. It can assist the chemist in starting-material search or for retrosynthetic analyses for either single structures or combinatorial libraries . The program automatically performs similarity searches in catalogues of available chemicals, defines strategic bonds and undertakes searches in reaction databases.

Collection of Computer Methods for Synthesis Design and Reaction Prediction J. Gasteiger, W.-D. Ihlenfeldt, P. Roese Recl. Trav. Chim. Pays-Bas 111 1992
Similarity Concepts for the Planning of Organic Reactions and Syntheses J. Gasteiger, W.-D. Ihlenfeldt, R. Fick, J. R. Rose J. Chem. Inf. Comput. Sci. 32 1992
Computer-Assisted Design of Syntheses for Heterocyclic Compounds R. Fick, W.-D. Ihlenfeldt, J. Gasteiger Heterocycles 40 1995
Computer-Assisted Synthesis and Reaction Planning in Combinatorial Chemistry J. Gasteiger, M. Pfoertner, M. Sitzmann, R. Hoellering, O. Sacher, T. Kostka, N. Karg Persp. Drug Discov.Design 20 2000
Other Programs ¶
Other programs for computer-aided synthetic planning are indicated in the following pages.

AIPHOS ¶
AIPHOS (Artificial Intelligence for Planning and Handling Organic Syntheses) was developed in 1988 at Toyohashi in Japan by S.I. Sasaki and K. Funatsu for the automated development of synthetic routes. The program automatically analyses the target compound, selects suitable starting material and reduces strategic sites and their corresponding reaction paths.

Computer-Assisted Organic Synthesis Design and Reaction Prediction System, 'AIPHOS' Funatsu, K.; Sasaki, S. I. Tetrahedron Comput. Methodol. 1 1988
Development of a Program for Construction of a Starting Material Library for AIPHOS, Koji Satoh, Shukou Azuma, Hiroko Satoh And Kimito Funatsu Journal of Chemical Software 4 1997
CAESA ¶
CAESA (Computer Assisted Estimation of Synthetic Accessibility) is a computer program introduced in 1991 by G.J. Myatt at the University of Leeds to automatically rank sets of molecules according to their ease of synthesis and to display the synthetic routes. It also assesses the development of viable synthetic approaches by identifying suitable, readily available starting materials. The software uses expert system technology to mimic the methods used by an experienced synthetic chemist to estimate the ease of synthesis of a particular compound.

AOCR ¶
AOCR (Automated Organic Chemical Reactions) is program developed by L. Ermanni (1993) based on a mathematical representation of organic synthesis, from the input of an organic compound. The program makes it possible to find all the syntheses of the target compound from the initial molecule given the reaction mechanisms. It also lists all the products of a reaction of the initial compounds.

SYSTEMATICHEM ¶
SystematiChem is an organic chemistry software program for retrosynthesis and synthesis development. The program provides an automated synthesis pathway generation tool that allows the user to enter the compound and set a few minor parameters for the solution generation process. It then generates complete synthesis pathways using only known, purchasable starting materials and documented reactions. After this process is completed, the generated routes of the solutions can be reviewed and evaluated by the chemist using a solution viewer program.

Databases for Organic Synthesis ¶
A huge amount of printed information, data and databases have accumulated over the years in the area of organic synthesis and are regularly updated. This well documented and well-structured information constitutes an essential form of assistance for all synthetic chemists. Some of the most important sources of information are outlined in some detail in the following pages.

Printed Information ¶
There is a vast number of textbooks, monograph series and articles that can be used by synthetic chemists as sources of information to find for example a detailed synthetic route, starting materials, conditions of a reaction, yields, catalysts, mechanisms etc... Examples of such printed information are shown below.

Methods of Organic Chemistry Thieme publisher 2001
Science of Synthesis: Houben-Weyl Methods of Molecular Transformations Georg Thieme Verlag, Stuttgart 2005
Organic Reactions Jossey-Bass publ. 61 volumes
Comprehensive Organic Chemistry Elsevier 1979
Comprehensive Organic Functional Group Transformations Elsevier 1995
Comprehensive Heterocyclic Chemistry I and II Pergamon Press 1984 - 1996
Comprehensive Natural Products Chemistry Pergamon Press 1999
Organic Syntheses
Encyclopedia of Reagents for Organic Syntheses Wiley 1995 +
Reagents for Organic Synthesis
Handbook of Reagents for Organic Syntheses Wiley 2000
Comprehensive Organic Transformations Wiley 1999
The Chemistry of Functional Groups Wiley Guide 1992
Compendium of Organic Synthetic Methods Wiley
Advanced Organic Chemistry Jossey-Bass 2001
Synthetic Methods of Organic Chemistry Karger Publ.
Comprehensive Organic Synthesis Pergamon Press 1991 +
The Logic of Chemical Synthesis Wiley 1989
Protective Groups in Organic Synthesis Wiley 1999
Contemporary Organic Synthesis The Royal Society of Chemistry 1994 +
Specialized Abstracting Services ¶
The Chemical Abstracts and Beilstein are two organizations that contributed substantially and for many years to the dissemination of scientific information in chemistry worldwide. Chemical Abstracts have existed for more than a century, whereas the Beilstein covers almost two centuries in chemistry. An outline of the resources provided by these two specialized abstracting services is given in the following pages.

Chemical Abstracts Services (CAS) ¶
CAS was founded in 1907 with the aim of monitoring, abstracting, and indexing the world's chemistry-related literature. In total, CAS has indexed and summarized chemistry-related articles from more than 40,000 scientific journals representing about 24 million documents and more than 15 million substances that are accessible online through CAS. The data produced by CAS are accessible to research scientists in different formats, including the printed CA, CA on CD, the STN international online network, the SciFinder and SciFinder-Scholar desktop research tools. CAS is a division of the American Chemical Society.

SciFinder ¶
SciFinder is used to access the CAS chemical information from a desktop with many types of searches. For reaction searches for example, once a structure is drawn, it is possible to refine the retrieved records with constraints such as the yield, the number of steps, the type of reaction etc...

Beilstein ¶
The Beilstein database contains fully searchable chemical structures and reactions, associated chemical and physical properties, and detailed pharmacological and ecological data. The chronological index began in 1771 and addresses three primary types of data: substances, reactions and citations. The Beilstein database is updated quarterly by Elsevier, a subsidiary of MDL Information Systems Inc.

Gmelin ¶
The Gmelin Database is the sister database to Beilstein, and has covered inorganic and organometallic compounds since 1772 to the present. It is used for graphic reaction searching. Gmelin is a product of MDL Information Systems.

Reaction Databases ¶
There are a number of databases that are dedicated to chemical reactions or organic syntheses and provide useful practical information from the point of view of the synthesis. Some examples of such specialized databases (free or commercial) are indicated here with their associated (clickable) internet address.

On-Line Resources ¶
If you are perplexed, have forgotten the content of a reaction referenced with a name (e.g. Stobbe condensation or Sakurai reaction) the on-line resources such as the "Organic Reaction Names" or the "Named Reactions in Organic Chemistry" might help you to rapidly recall it and view some examples. A vast number of resources are available on-line for the synthetic chemists; the list indicated below indicates only some of these. Portals such as the American Chemical Society or Organic Chemistry Worldwide Resources provide additional useful links.

Patent Databases ¶
In the development phase of a drug, patent databases provide useful information on its synthesis which has not been published in the scientific articles or poster forms. Moreover, when engaged in a discovery project, it is important to know what types of structures are well covered by the competition. In the following pages some important patent databases are presented.

US Patents: USPTO ¶
The United States Patent and Trademark Office (USPTO) provides free web access to its database of issued patents and published applications. Quick and advanced searches are possible with fields such as words, inventor name, inventor country, assignee name and country, issue date, examiner, patent number etc... The USPTO is an agency of the US Department of Commerce and the database is updated weekly.
European Patents: EPOLINE ¶
The European Patent Register On-Line (EPOLINE) provides information on all European and Euro-PCT patent applications. Records include bibliographic data, legal status information but no description of the invention (claims or abstracts). Files are updated daily and information is available to online users 3-5 days after the action date.

Other Patent Databases ¶
Other patent databases exist, some of which are indicated below. In particular, the IBM's Patent Server is a public service providing a different patent database from the US Patent abstracts with as service similar to that of the USPTO. Also, the Japanese Patent Office has a searchable database of Japanese patent abstracts, which include the patent number, title, inventor, company, and abstract of the patent.

Copyright © 2024 drugdesign.org
- Even more »
Account Options

- Try the new Google Books
- Advanced Book Search
- Oxford University Press
- Barnes&Noble.com
- Books-A-Million
- Find in a library
- All sellers »

Get Textbooks on Google Play
Rent and save from the world's largest eBookstore. Read, highlight, and take notes, across web, tablet, and phone.
Go to Google Play Now »
Selected pages
Common terms and phrases
About the author (2015), bibliographic information.
Click through the PLOS taxonomy to find articles in your field.
For more information about PLOS Subject Areas, click here .
- Drug synthesis
- Pharmaceutical processing technology
- Get an email alert for Drug synthesis
- Get the RSS feed for Drug synthesis
Showing 1 - 13 of 14
View by: Cover Page List Articles
Sort by: Recent Popular
Indenyl-thiazole and indenyl-formazan derivatives: Synthesis, anticancer screening studies, molecular-docking, and pharmacokinetic/ molin-spiration properties
Ghaidaa H. Alfaifi, Thoraya A. Farghaly, Magda H. Abdellattif
Hospital at home: A systematic review of how medication management is conceptualised, described and implemented in practice—A study protocol
Sophie McGlen, Clare Crowley, [ ... ], Rosemary H. M. Lim
Comparative statistical analysis of the release kinetics models for nanoprecipitated drug delivery systems based on poly(lactic-co-glycolic acid)
Nathaly S. Heredia, Karla Vizuete, [ ... ], Alexis Debut
Inhibition and assessment of the biophysical gating properties of GluA2 and GluA2/A3 AMPA receptors using curcumin derivatives
Mohammad Qneibi, Othman Hamed, [ ... ], Rola Al-Kerm
Synthesis of ordered lamellar supermicroporous silica with rigid neutral and long-chain cationic composite templating route
Shangxing Chen, Peng Wang, [ ... ], Zongde Wang
Guidelines for safe handling of hazardous drugs: A systematic review
Mari A. Bernabeu-Martínez, Mateo Ramos Merino, [ ... ], Javier Sanz-Valero
in vitro release of 5-fluorouracil">pH responsive N-succinyl chitosan/Poly (acrylamide-co-acrylic acid) hydrogels and in vitro release of 5-fluorouracil
Shahid Bashir, Yin Yin Teo, [ ... ], K. Ramesh
Ordered lamellar supermicroporous titania templating by rosin-derived quaternary ammonium salt
Fei Song, Peng Wang, [ ... ], Guorong Fan
Time Savings with Rituximab Subcutaneous Injection versus Rituximab Intravenous Infusion: A Time and Motion Study in Eight Countries
Erwin De Cock, Persefoni Kritikou, [ ... ], Tim Waterboer
A New Application of Parallel Synthesis Strategy for Discovery of Amide-Linked Small Molecules as Potent Chondroprotective Agents in TNF-α-Stimulated Chondrocytes
Chia-Chung Lee, Yang Lo, [ ... ], Hsu-Shan Huang
Dextran Nanoparticle Synthesis and Properties
Iga Wasiak, Aleksandra Kulikowska, [ ... ], Tomasz Ciach
A Nano-MgO and Ionic Liquid-Catalyzed ‘Green’ Synthesis Protocol for the Development of Adamantyl-Imidazolo-Thiadiazoles as Anti-Tuberculosis Agents Targeting Sterol 14α-Demethylase (CYP51)
Sebastian Anusha, Baburajeev CP, [ ... ], Kanchugarakoppal S. Rangappa
A Systematic Review of Mapping Strategies for the Sonification of Physical Quantities
Gaël Dubus, Roberto Bresin
Connect with Us
- PLOS ONE on Twitter
- PLOS on Facebook
Introduction to Click Chemistry
A "click" away from discovery.
The traditional process of drug discovery based on natural secondary metabolites has often been slow, costly, and labor-intensive. Even with the advent of combinatorial chemistry and high-throughput screening in past decades, the generation of leads is dependent on the reliability of the individual reactions to construct the new molecular framework.
Click Chemistry Mechanism
Click chemistry is a newer approach to the synthesis of drug-like molecules that can accelerate the drug discovery process by utilizing a few practical and reliable reactions. Sharpless and coworkers defined what makes a click reaction as one that is wide in scope and easy to perform, uses only readily available reagents, and is insensitive to oxygen and water. In fact, in several instances, water is the ideal reaction solvent, providing the best yields and highest rates. Reaction work-up and purification uses benign solvents and avoids chromatography. 1
Click Chemistry Reaction Processes
- Simple to perform
- Wide in scope
- High yielding
- Stereospecific
- Adheres to the 12 Principles of Green Chemistry by generating harmless byproducts, removable by nonchromatographic methods
Click Chemistry Reaction Characteristics 1
- Simple reaction conditions
- Readily and easily available starting materials and reagents
- Use of no solvent, a benign solvent (such as water), or one that is easily removed
- Simple product isolation
- Product should be stable under physiological conditions
Click chemistry involves the use of a modular approach and has important applications in the fields of drug discovery, combinatorial chemistry, target-templated in situ chemistry, and DNA research. 1
Continue learning about click chemistry in drug discovery .
Network error: Failed to fetch
- Triazolylphenylglyoxal Reagents: Arginine-Directed Bioconjugation
- SAFC Biologic APIs and Conjugation Experts
- Click Chemistry in Drug Discovery
- Copper-Free Click Chemistry
- GlycoProfile™ Azido Sugars
- Guanylation of Amines by Bis(tert-butoxycarbonyl)thiopseudourea, Polymer-Bound
- Polybrene® Technical Bulletin
- Safe Manufacture of Antibody Drug Conjugates
To continue reading please sign in or create an account.
Research. Development. Production.
We are a leading supplier to the global Life Science industry with solutions and services for research, biotechnology development and production, and pharmaceutical drug therapy development and production.
© 2024 Merck KGaA, Darmstadt, Germany and/or its affiliates. All Rights Reserved.
Reproduction of any materials from the site is strictly forbidden without permission.
- English - EN
- Español - ES
An AI leap into chemical synthesis
EPFL scientists introduce ChemCrow, a large language model-based AI system that revolutionizes chemistry by integrating 18 advanced tools for tasks like organic synthesis and drug discovery. ChemCrow streamlines complex processes in chemical research, making it more efficient for experts and novices alike.
Chemistry, with its intricate processes and vast potential for innovation, has always been a challenge for automation. Traditional computational tools, despite their advanced capabilities, often remain underutilized due to their complexity and the specialized knowledge required to operate them.
Now, researchers with the group of Philippe Schwaller at EPFL, have developed ChemCrow, an AI that integrates 18 expertly designed tools, enabling it to navigate and perform tasks within chemical research with unprecedented efficiency. "You might wonder why a crow?" asks Schwaller. "Because crows are known to use tools well."
ChemCrow was developed by PhD students Andres Bran and Oliver Schilter (EPFL, NCCR Catalysis) in collaboration with Sam Cox and Professor Andrew White at (FutureHouse and University of Rochester).
ChemCrow is based on a large language model (LLMs), such as GPT-4, enhanced by LangChain for tool integration, to autonomously perform chemical synthesis tasks. The scientists augmented the language model with a suite of specialized software tools already used in chemistry, including WebSearch for internet-based information retrieval, LitSearch for scientific literature extraction, and various molecular and reaction tools for chemical analysis.
By integrating ChemCrow with these tools, the researchers enabled it to autonomously plan and execute chemical syntheses, such as creating an insect repellent and various organocatalysts, and even assist in discovering new chromophores, substances fundamental to dye and pigment industries.
What sets ChemCrow apart is its ability to adapt and apply a structured reasoning process to chemical tasks. "The system is analogous to a human expert with access to a calculator and databases that not only improve the expert's efficiency, but also make them more factual -- in the case of ChemCrow, reducing hallucinations," explains Andres Camilo Marulanda Bran, the study's first author.
ChemCrow receives a prompt from the user, plans ahead how to solve the task, selects the relevant tools, and iteratively refines its strategy based on the outcome(s) of each step. This methodical approach ensures that ChemCrow doesn't only work off theory but is also grounded in practical application for real-world interaction with laboratory environments.
By democratizing access to complex chemical knowledge and processes, ChemCrow lowers the barrier to entry for non-experts while augmenting the toolkit available to veteran chemists. This can accelerate research and development in pharmaceuticals, materials science, and beyond, making the process more efficient and safer.
The group of Philippe Schwaller is part of the new EPFL AI Center , with more than forty other laboratories, leading the way towards trustworthy, accessible and inclusive AI.
- Organic Chemistry
- Inorganic Chemistry
- Biochemistry
- Computer Modeling
- Mathematical Modeling
- Organic chemistry
- Global climate model
- Mathematical model
- Artificial neural network
- Computer simulation
- Resonance (chemistry)
Story Source:
Materials provided by Ecole Polytechnique Fédérale de Lausanne . Original written by Nik Papageorgiou. The original text of this story is licensed under Creative Commons CC BY-SA 4.0 . Note: Content may be edited for style and length.
Journal Reference :
- Andres M. Bran, Sam Cox, Oliver Schilter, Carlo Baldassari, Andrew D. White, Philippe Schwaller. Augmenting large language models with chemistry tools . Nature Machine Intelligence , 2024; DOI: 10.1038/s42256-024-00832-8
Cite This Page :
Explore More
- 'Major' Ancient Migration to Timor Island
- Ancient Viral DNA: Psychiatric Disorders
- New AI Accurately Predicts Fly Behavior
- Extreme Impacts: Metals Stronger When Heated
- Rare Earth Element's Secrets Exposed
- Origin of Sun's Magnetic Field
- Measuring Spin of Supermassive Black Hole
- 'Cell Cannibalism' Across Tree of Life
- Very Low Emission Concrete at Scale
- Kangaroos: Fearing Human 'Super Predator'
Trending Topics
Strange & offbeat.
Advertisement
Strobilurin X acts as an anticancer drug by inhibiting protein synthesis and suppressing mitochondrial respiratory chain activity
- Open access
- Published: 20 May 2024
- Volume 15 , article number 177 , ( 2024 )
Cite this article
You have full access to this open access article

- Kenji Takahashi 1 , 2 ,
- Tomoya Tanaka 3 ,
- Atsushi Ishihara 4 &
- Toshio Ohta 1 , 2
Explore all metrics
Strobilurins act as antifungal agents by inhibiting the mitochondrial respiratory chain. The cytotoxic activity of strobilurins, focusing on its anticancer activities, has been reported. However, the mechanisms involved in these activities remain unclear.
The cytotoxic effects of strobilurin X isolated from the mycelium of Mucidula. venosolamellata were examined in human cancer cell lines (A549 and HeLa) and normal fibroblasts (WI-38).
Strobilurin X significantly decreased the viability of A549 and HeLa cells compared to that in the WI-38 cells after 48 h of exposure. The EC 50 values for cytotoxicity in the A549, HeLa, and WI-38 cells were 3.4, 5.4, and 16.8 μg/mL, respectively. Strobilurin X inhibited the mitochondrial respiratory chain and enhanced the release of lactate in the A549 cells. The IC 50 value of strobilurin X against the mitochondrial respiratory chain complex III activity was 139.8 ng/mL. The cytotoxicity induced by strobilurin X was not completely rescued after adding uridine, methyl pyruvate, or N -acetyl cysteine. Furthermore, pharmacological approaches demonstrated that strobilurin X failed to modulate the mitogen-activated protein kinase family and phosphoinositide 3-kinase-Akt pathways; alternatively, it suppressed protein synthesis independent of uridine.
Strobilurin X induced cytotoxicity by blocking the mitochondrial respiratory chain and suppressing protein synthesis. These findings may aid in the development of novel anticancer drugs using strobilurins.
Similar content being viewed by others

Therapeutic potential of pyrrole and pyrrolidine analogs: an update

Understanding the mechanism of action of protease inhibitors in controlling the growth of the Candida Genus: potential candidates for development of new antifungal molecules

Molecular and therapeutic insights of rapamycin: a multi-faceted drug from Streptomyces hygroscopicus
Avoid common mistakes on your manuscript.
1 Introduction
Strobilurins are antifungal pesticides widely used in agriculture across the world [ 1 , 2 ]. Strobilurins A and B were initially isolated from the mycelium of the basidiomycete Strobilurus tenacellus [ 3 ]. Mucidin extracted from Oudemansiella mucida [ 4 ] was structurally identical to strobilurin A [ 5 ]. Strobilurins exert antifungal effects by inhibiting the transfer of an electron in the mitochondrial respiratory chain at complex III [ 5 , 6 , 7 , 8 , 9 ]. Several types of strobilurins have been isolated and identified from various mushrooms and synthesized for the development of pesticides [ 1 , 10 ]. Strobilurin X was detected in the culture medium of Oudemansiella mucida and showed the higher antifugal activity than strobilurin A [ 11 , 12 ].
Cancer is the most common morbidity, leading to a growing demand for the development of novel drugs. Despite many studies on the anticancer effects of various mushroom extracts, the findings have not been exploited for clinical use [ 13 ]. The mitochondrial respiratory chain is one of many therapeutic targets for cancer treatments because it is essential for cell proliferation [ 14 ]. In one study, the deletion of complex III, one of the mitochondrial respiratory chain components, abolished tumor cell growth, resulting in the amplification of the tumor engrafted in mice [ 15 ]. However, no compounds inhibiting complex III have been successfully used to develop anticancer drugs. Strobilurins inhibit the activity of complex III by suppressing cytochrome bc1 [ 5 , 13 ]; thus, they may be useful for the development of compounds that can inhibit cancer cell proliferation.
The aim of this study was to examine the cytotoxic effects of strobilurin X, isolated from Mucidula venosolamellata, on cancer cells. Strobilurin X significantly suppressed cell proliferation and inhibited the mitochondrial respiratory chain complex III activity. However, the cytotoxicity was not solely attributed to the suppression of complex III, indicating that strobilurin X had alternative targets for its cytotoxic effect. Further experiments revealed that strobilurin X inhibited protein synthesis. These results suggested that strobilurin X exerts anticancer effects and may serve as a lead compound for developing new anticancer drugs.
2.1 Preparation of strobilurin X
Strobilurin X was extracted from M. venosolamellata (TUFC 30333, Fungus/Mushroom Resource and Research Center, Faculty of Agriculture, Tottori University). M. venosolamellata was cultured in 5 L of malt extract media for 30–60 days. The culture filtrate was extracted using ethyl acetate (1 L × 3) and fractionated by silica gel column chromatography (acetone-hexane, 10% stepwise from 0 to 30% acetone). The 10% acetone fraction was further fractionated by silica gel column chromatography (ethyl acetate-hexane, 10% stepwise elution from 0 to 30%). Strobilurin X was purified from the 20% ethyl acetate fraction via preparative high-performance liquid chromatography using a Cosmosil 5C18 ARII (10 × 250 mm, Nacalai Tesque, Kyoto, Japan) column and 65% acetonitrile–water as solvent (flow rate: 3 mL/min, ultraviolet detection: 254 nm, [Shimadzu 10A system, Kyoto, Japan]). The identity of the compound was confirmed by nuclear magnetic resonance and mass spectra according to our recent report [ 16 ].
2.2 Cell culture
Lung and cervical cancer cell lines (A549 and HeLa, respectively) and normal lung fibroblasts (WI-38) were purchased from Riken Bioresource Center (Tsukuba, Japan) and cultured in Dulbecco’s modified Eagle medium (D6429; Sigma-Aldrich, Tokyo, Japan) supplemented with 10% bovine fatal serum, 100 U/mL streptomycin (Meiji Seika Co. Ltd, Tokyo, Japan) and 100 μg/mL penicillin (Meiji Seika Co. Ltd).
2.3 Detection of cell viability using the WST-8 assay
Cell viability was assessed using Cell Counting Kit-8 (WST-8; Dojin, Tokyo, Japan). In brief, the cells (5 × 10 3 cells/well) were prepared in a 96-well culture plate the day before the experiment. Various concentrations of strobilurin X were applied for 48 h, and the cell viability was evaluated by measuring the absorbance with a microplate spectrometer (Sunrise ™ Fuji film Wako Pure Chemicals, Osaka, Japan) at 450 nm between 1 and 4 h after the addition of the WST-8 reagent to the culture medium in each well. The ratio of each data set to that of the vehicle was calculated.
2.4 Quantitation of lactate production from the cells
The amount of lactate produced through glycolysis was measured using the Lactate Assay Kit-WST (Dojin). The cells (10 4 cells/well) were prepared in the 96-well culture plate the day before the experiment and incubated with various concentrations of strobilurin X for 48 h. A portion of each conditioned medium was transferred to a new 96-well culture plate and mixed with a working solution, according to the operating instructions. After incubation for 30 min at 37 °C, the absorbance of each sample was measured at 450 nm. The amount of lactate produced by the cells under each condition was calculated using a standard curve.
2.5 Measurement of the mitochondrial respiratory chain complex III activity in cell-free system
The mitochondrial respiratory chain complex III activity was measured using the MitoCheck ® Complex II/III Activity Assay Kit (Cayman Chemical, Ann Arbor, MI, USA) according to recent reports [ 17 , 18 ]. In brief, the reduction of cytochrome c by complex III was detected at 550 nm using a spectrophotometer (Genesys 10S UV–Vis; ThermoFisher Scientific, Tokyo, Japan). A working solution of strobilurin X was mixed with cytochrome c solution and bovine heart mitochondria, and the absorbance was measured every 30 s for 10 min. The activity of complex III was calculated as the ratio of the rate of change in absorbance in each sample to that in the vehicle.
2.6 Detection of the protein synthesis activity
The protein synthesis activity was assessed using the Cayman’s Protein Synthesis Assay Kit (Cayman Chemical). In brief, the cells (3 × 10 3 cells/well) were prepared in a 96-well culture plate with a black bottom (Thermo Fisher Scientific) the day before the experiment. The cells were pretreated with strobilurin X for 30 min and labeled with O -Propargyl-puromycin (OPP) for 2 h. The amount of 5-carboxyfluorescein (FAM) bound to OPP was determined by measuring the fluorescence at 535 nm with excitation at 488 nm using a fluorescent microscope (BZ-X810, Keyence, Osaka, Japan).
2.7 Measurement of intracellular reactive oxygen species (ROS) production
After exposure to strobilurin X, the cells harvested by trypsinization were washed with phosphate-buffered saline (PBS) and loaded with chloromethyl-2ʹ,7ʹ—dichlorodihydrofluorescein diacetate (CM-H 2 DCFDA; 40 μM; C6827, Thermo Fisher Scientific) for 30 min at 37 °C. Subsequently, the cells were washed with PBS and analyzed using a flow cytometer (FACS Aria, BD Japan, Tokyo, Japan).
2.8 Chemicals
Uridine (solvent, stock concentration; PBS, 200 mg/ml), methyl pyruvate (PBS, 110 mg/mL) and N -acetyl cysteine (1 M NaOH, 1 M) are purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan). PD98059 (dimethylsulfoxide [DMSO], 50 mM), U0126 (DMSO, 20 mM), LY2940001 (DMSO 5 mM), JNK inhibitor II (DMSO, 10 mM) and SB203580 (DMSO, 10 mM) were obtained from Calbiochem (Sigma-Aldrich Japan, Tokyo, Japan).
2.9 Statistical analysis
All analyses were conducted using the SPSS Statistics software (IBM Japan, Tokyo, Japan). Student’s or Welch’s t -test was used for two-group comparisons, and one-way analysis of variance followed by the Games-Hawell test was used for multiple-group comparisons. A p-value of < 0.05 was considered significant.
3.1 Cytotoxicity induced by strobilurin X
Strobilurins have been reported to induce cell death [ 19 , 20 ]. Therefore, the cytotoxic effect of strobilurin X was examined in the current study. Strobilurin X significantly decreased the viability of the A549 and HeLa cells at 48 h in a concentration-dependent manner. The EC 50 values of strobilurin X in the A549 and HeLa cells were 3.4 ± 0.2 and 5.4 ± 0.3 μg/mL, respectively (Figs. 1 A and B ). Although a cytotoxic effect was observed in the WI-38 cells, the EC 50 value in these cells (16.8 ± 3.4 μg/mL) was higher than those in the two cancer cell lines (Fig. 1 C). These results suggest that strobilurin X has a relatively selective cytotoxic effect on cancer cells.

Cytotoxicity induced by strobilurin X. A Chemical structure of strobilurin X. Relationships between viability and the concentrations of strobilurin X at 48 h in the A549 B , HeLa C , and WI-38 D cells. Cell viability is indicated as the ratio of each value to that of the vehicle. Symbols with vertical lines show the mean ± the standard error of the mean (SEM) in three separate experiments
3.2 Involvement of blockade of mitochondrial respiratory chain complex III in strobilurin X-induced cytotoxicity
A549 cells were used to evaluate the detail cytotoxic effects and mechanisms of strobilurin X. Inhibition of the mitochondrial respiratory chain enhances the glycolytic system [ 21 ]; therefore, the amount of lactate effluxed by glycolysis from the A549 cells was therefore measured in the current study. Strobilurin X increased lactate release at 48 h (Fig. 2 A) and inhibited the activity of the mitochondrial respiratory chain complex III in a concentration-dependent manner. The IC 50 of strobilurin X was 139.8 ± 32.0 ng/mL; however, this value was significantly lower than the concentration showing cytotoxicity in A549 (Fig. 2 B). These results clearly showed that strobilurin X inhibited mitochondrial respiratory chain complex III, although the experimental setting was different between the measurement of complex III activity and cytotoxicity. Future experiments using extracts from used cells may reveal details.

The effects of strobilurin X on mitochondrial respiratory chain activity. A The amount of extracellular lactate from A549 cells after stimulation with strobilurin X for 48 h. The column with a vertical line shows the mean ± SEM in three separate experiments. B The relationship between the mitochondrial respiratory chain complex III activity (○) or cell viability (●) and the concentrations of strobilurin X. Complex III activity is indicated as the percentage of the rate of change in each activity to that of the vehicle. The cell viability data is transcribed from Fig. 1 A. Symbols with vertical lines show the mean ± SEM in three experiments. **p < 0.01 vs. vehicle using the Games-Hawell test
As shown in Fig. 3 A, uridine (50 μg/mL) significantly reduced the cytotoxicity of strobilurin X, thus indicating that pyrimidine biosynthesis was inhibited by strobilurin X through the mitochondrial respiratory chain. Uridine rescued the cytotoxicity of strobilurin X (6.25 μg/mL) in a concentration-dependent manner, but did not completely overcome the cytotoxic effect even at 200 μg/mL (Fig. 3 B). Pyruvate is also known to reduce the cytotoxicity induced by mitochondrial respiratory chain dysfunction [ 15 , 22 ]. As shown in Fig. 3 C, methyl pyruvate did not attenuate the strobilurin X-induced cytotoxic effect. These results suggest that other targets, besides the inhibition of the mitochondrial respiratory chain complex III, may be involved in the cytotoxic action of strobilurin X.

Effects of uridine on the strobilurin X-induced cytotoxicity in A549 cells. A Relationships between the cell viability and concentrations of strobilurin X at 48 h without (●) and with uridine (○; 50 μg/mL). Cell viability is indicated as the ratio of each value to that of the vehicle without uridine. Symbols with vertical lines show the mean ± SEM in three separate experiments. B , C Relationship between the cell viability induced by the vehicle (open columns) and strobilurin X (closed columns, 6.25 μg/mL) and the concentrations of uridine B or methyl pyruvate (C; 110 μg/mL) at 48 h. Columns with vertical lines show the mean ± SEM in three separate experiments. *p < 0.05 vs. the vehicle with uridine using Student’s t -test or Welch’s t -test
3.3 Inhibition of protein synthesis by strobilurin X
Among the cell growth signals, PD98059, U0126, mitogen activated protein kinase (MAPK)/extracellular signal-regulated kinase (Erk) kinase (MEK) inhibitors, and LY2940001, a phosphoinositide 3-kinase (PI3K) inhibitor, failed to modulate the strobilurin X (6.25 μg/mL)-induced cytotoxicity. Likewise, among the cell death signals, JNK inhibitor II and SB203580, a p38 MAPK inhibitor, did not affect the strobilurin X-induced cell death (Fig. 4 ). These results suggest that the cell growth and cell death signals are not related to the cytotoxic effect of strobilurin X.

Effects of inhibitors of cell proliferation and death signals on the cytotoxicity induced by strobilurin X in the A549 cells. Cell viability is indicated as the ratio of each value to that of the vehicle without inhibitors at 24 h (open columns, vehicle; closed columns, strobilurin X [6.25 μg/mL]). Veh, vehicle; PD, 10 μM PD98059; U01, 10 μM U0126; LY, 5 μM LY2940004; JNKi, 5 μM JNK inhibitor; SB, 10 μM SB203950. Columns with vertical lines show the mean ± SEM in three separate experiments
Various anticancer compounds have generated intracellular ROS, which induced cellular damage [ 23 , 24 ]. The number of A549 cells emitting stronger fluorescence for the ROS indicator dichlorodihydrofluorescein (DCF) increased to the maximum level after 12 h of exposure to strobilurin X, although the mean fluorescence intensity was lower than that of number of hydrogen peroxide (0.5 mM) for 1 h (Fig. 5 A and B ). The strobilurin X-induced high fluorescent intensity levels were maintained up to 24 h (Fig. 5 B). However, N -acetyl cysteine, a potent ROS scavenger, failed to attenuate the strobilurin X-induced cytotoxicity in A549 cells (Fig. 5 C). These results suggest that strobilurin X induces the production of intracellular ROS, but its effect is not essential for the cytotoxicity.

ROS production is not involved in the cytotoxicity induced by strobilurin X. A Typical histograms showing the cell numbers with DCF fluorescence after exposure to vehicle (12 h), strobilurin X (6.25 μg/mL, 12 h) and hydrogen peroxide (H 2 O 2 , 0.5 mM, 1 h). The mean fluorescence intensity (MFI) was calculated using the BD FACSDiva ™ software. B The time course of ROS production, indicated as the ratio of each MFI to that of the vehicle at each time point after exposure to strobilurin X. Symbols with vertical lines show the mean ± SEM in three separate experiments. The dashed line shows a baseline equal to the level in the vehicle. *p < 0.05 vs. the vehicle using Student’s t -test. C The effect of N -acetyl cysteine (NAC; 1 mM) on strobilurin X-induced cytotoxicity in A549 cells at 48 h. Cell viability is indicated as the ratio of each value to that of the vehicle without NAC (open columns, vehicle; closed columns, strobilurin X [6.25 μg/mL]). Cells were pretreated with NAC 30 min before exposure to strobilurin X. Columns with vertical lines show the mean ± SEM in three separate experiments
Recently, the link between protein synthesis inhibition and anticancer activity has been focused on certain compounds [ 25 ]. Protein synthesis is indispensable in cell proliferation; hence, we evaluated the protein synthesis activity using OPP, which was labeled on the C-terminus of nascent polypeptide chains [ 26 ]. Strobilurin X decreased the cell fluorescent intensity labeled by protein synthesis for 2 h in A549 cells, indicating that strobilurin X inhibits protein synthesis (Fig. 6 A). Uridine failed to block the inhibition of protein synthesis by strobilurin X (Fig. 6 B). These results suggest that strobilurin X can inhibit protein synthesis and mitochondrial respiratory chain complex III.

Effect of strobilurin X on protein synthesis in A549 cells. A Typical fluorescent microscope images of the protein synthesis activity 2 h after exposure to strobilurin X (upper, vehicle; lower, 25 μg/mL of strobilurin X). White scale bars: 50 μm. B The fluorescent amount in each well without (vehicle) or with uridine (50 μg/mL) (open columns, vehicle; closed columns, 25 μg/mL of strobilurin X). Columns with vertical lines show the mean ± SEM in three separate experiments. *p < 0.05 vs. the vehicle using the Games-Hawel test. NS not significant
4 Discussion
In this study, we found that the cytotoxic effects of strobilurin X (isolated from M. venosolamellata) on cancer cells were greater than those on normal fibroblasts. Moreover, strobilurin X inhibited protein synthesis and mitochondrial respiratory chain complex III in the lung cancer cell line.
The mitochondrial respiratory chain is essential for generating the intracellular concentration of ATP. It is considered as a target for anticancer effects owing to its ability to regulate the metabolism and proliferation of cancer cells [ 27 ]. The oxidation of dihydroorotate dehydrogenase by complex III produces orotates, which lead to pyrimidine biosynthesis [ 28 ]. The depletion of complex III inhibits de novo pyrimidine synthesis and induces cytotoxicity in cancer cells [ 15 ]. A commercially available strobilurin pesticide, azoxystrobin, was reported to induce cytotoxicity, which was entirely rescued by uridine in myelogenous leukemia HL-60RG cells [ 29 ]. In the current study, strobilurin X-induced cytotoxicity was partially abolished by uridine, whereas methyl pyruvate did not have any effect in attenuating the strobilurin X-induced cytotoxicity (Fig. 3 ). Therefore, we hypothesized that other mechanisms, apart from mitochondrial respiratory complex III inhibition, were involved in the cytotoxicity of A549 cells.
Strobilurin X was found to directly suppress protein synthesis in this study (Fig. 6 ). Strobilurins A and B were reported to attenuate the incorporation of radioactive leucine in Ehrlich carcinoma ascites cells [ 3 ]. Direct evidence for the inhibition of protein synthesis by strobilurin X was seen for the first time in the present study. The chemical structure of strobilurin X is different from that of strobilurin A in terms of the presence of a methoxy group at the para position of its benzene ring; it is different from that of strobilurin B at the position of the methoxy group and the Cl substituents and significantly different from that of azoxystrobin [ 1 , 3 , 11 ]. Therefore, the inhibition of protein synthesis may be dependent on the structure of the strobilurins. Nonetheless, future studies identifying the molecular targets and clarifying the structure–activity relationship in protein synthesis are warranted.
Strobilurins are generally used as pesticides but are currently being evaluated as anticancer drugs [ 30 ]. Interestingly, azoxystrobin induced cytotoxicity by inhibiting the phosphorylation of PI3K/Akt and Erk [ 31 ]. However, the inhibitors of these molecules did not affect the cytotoxic action of strobilurin X in the present study. The cytotoxic activity of strobilurin X may not be related to these cell growth signals in A549 cells.
Natural compounds such as phytochemicals often generate ROS, which evokes cytotoxicity [ 23 , 24 ]. Strobilurin X induced ROS generation in the current study. It may induce the generation of ROS in mitochondria due to the inhibition of mitochondrial respiratory chain complex III [ 32 ]. However, the potent antioxidant N -acetyl cysteine failed to suppress the strobilurin X-induced cytotoxicity in this study (Fig. 5 ). Similarly, mito-TEMPO, an intra-mitochondria ROS scavenger, also failed to block the strobilurin X-induced cytotoxicity (data not shown). Furthermore, strobilurin X did not induce the generation of ROS in HeLa cells despite its cytotoxicity (Fig. 1 and data not shown). Therefore, the generation of ROS by strobilurin X may not be essential for its cytotoxic action. Alternatively, the amount of ROS generated by strobilurin X may not be sufficient to kill A549 cells.
In summary, the inhibition of mitochondrial respiratory chain complex III and protein synthesis was found to be related to the cytotoxic effects of strobilurin X in A549 cells. Strobilurin-related compounds are widely used as pesticides; thus, the drug repositioning may be expected. The findings of this study indicate that strobilurin X may be considered for the development of novel anticancer drugs.
Data availability
All data included in this study are available from the corresponding author upon appropriate request.
Code availability
Not applicable.
Abbreviations
Dichlorodihydrofluorescein
Dimethylsulfoxide
Extracellular signal-regulated kinase
5-Carboxyfluorescein
2ʹ,7ʹ—Dichlorodihydrofluorescein diacetate
Modulate mitogen-activated protein kinase
Mitogen activated protein kinase/extracellular signal-regulated kinase kinase
N -Acetyl cysteine
O -Propagyl-puromycin
Phosphate-buffered saline
Phosphoinositide 3-kinase
Reactive oxygen species
Standard error of the mean
Balba H. Review of strobilurin fungicide chemicals. J Environ Sci Health B. 2007;42:441–51.
Article CAS PubMed Google Scholar
Feng Y, Huang Y, Zhan H, Bhatt P, Chen S. An overview of strobilurin fungicide degradation: current status and future perspective. Front Microbiol. 2020;11:389.
Article PubMed PubMed Central Google Scholar
Anke T, Oberwinkler F. The strobilurins—new antifungal antibiotics from the basidiomycete Strobilurus tenacellus . J Antibiot. 1977;30:806–10.
Article CAS Google Scholar
Musílek V, Černă J, Šašek V, Semerdžieva M, Vondráček M. Antifungal antibiotic of the basidiomycete Oudemansiella mucida I isolation and cultivation of a producing strain. Folia Microbiol. 1969;14:377–87.
Article Google Scholar
Von Jagow G, Gribble GW, Trumpower BL. Mucidin and strobilurin A are identical and inhibit electron transfer in the cytochrome bc1 complex of the mitochondrial respiratory chain at the same site as myxothiazol. Biochemistry. 1986;25:775–80.
Šubík J, Behúň M, Šmigáň P, Musílek V. Mode of action of mucidin, a new antifungal antibiotic produced by the basidiomycete Oudemansiella mucida . Biochimica Biophysica Acta. 1974;343:363–70.
Trowitzsch W, Reifenstahl G, Wray V, Gerth K. Myxothiazol: a reversible blocker of the cell cycle. J Antibiot. 1980;33:1480–90.
Becker WF, von Jagow G, Anke T, Steglich W. Oudemansin, strobilurin A, strobilurin B and myxothiazol: new inhibitors of the bc 1 segment of the respiratory chain with an E-β-methoxyacrylate system as common structural element. FEBS Lett. 1981;132:329–33.
Musso L, Fabbrini A, Dallavalle S. Natural compound-derived cytochrome bc1 complex inhibitors as antifungal agents. Molecules. 2020;25:4582.
Article CAS PubMed PubMed Central Google Scholar
Sauter H, Steglich W, Anke T. Strobilurins: evolution of a new class of active substances. Angew Chem Int Ed. 1999;38:1328–49.
Vondráček M, Vondráčková J, Sedmera P, Musílek V. Another antibiotic from the basidiomycete Oudemansiella mucida . Collection Czechoslovak Chem Commun. 1983;48:1508–12.
Kettering M, Sterner O, Anke T. Antibiotics in the chemical communication of fungi. Naturforsch. 2004. https://doi.org/10.1515/znc-2004-11-1209 .
Panda SK, Sahoo G, Swain SS, Luyten W. Anticancer activities of mushrooms: a neglected source for drug discovery. Pharmaceuticals. 2022;5:176.
Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32:341–52.
Martínez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, Kihshen H, Reczek CR, Weinberg SE, Gao P, Steinert EM, Piseaux R, Budinger GRS, Chandel NS. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature. 2020;585:288–92.
Tanaka T, Takahashi K, Inoue Y, Endo N, Shimoda E, Ueno K, Ichiyanagi T, Ohta T, Ishihara A. Inhibition of melanoma cell proliferation by strobilurins isolated from mushrooms and their synthetic analogues. Biosci Biotechnol Biochem. 2024;88:389–98.
Article PubMed Google Scholar
Zhang C, Liu Z, Bunker E, Ramirez A, Lee S, Peng Y, Tan A-C, Eckhardt SG, Chapnick DA, Liu X. Sorafenib targets the mitochondrial electron transport chain complexes and ATP synthase to activate the PINK1–Parkin pathway and modulate cellular drug response. J Biol Chem. 2017;292:15105–20.
Heishima K, Sugito N, Soga T, Nishikawa M, Ito Y, Honda R, Kuranaga Y, Sakai H, Ito R, Nakagawa T, Ueda H, Akao Y. Petasin potently inhibits mitochondrial complex I–based metabolism that supports tumor growth and metastasis. J Clin Invest. 2021;131: e139933.
Lee S, Kwon OS, Lee CS, Won M, Ban HS, Ra CS. Synthesis and biological evaluation of kresoxim-methyl analogues as novel inhibitors of hypoxia-inducible factor (HIF)-1 accumulation in cancer cells. Bioorg Medicin Chem Lett. 2017;27:3026–9.
Shi XK, Bian XB, Huang T, Wen B, Zhao L, Mu HX, Fatima S, Fan BM, Bian ZX, Huang LF, Lin CY. Azoxystrobin induces apoptosis of human esophageal squamous cell carcinoma KYSE-150 cells through triggering of the mitochondrial pathway. Front Pharmacol. 2017;8:277.
Lan J, Cadassou O, Corbet C, Riant O, Feron O. Discovery of mitochondrial complex I inhibitors as anticancer and radiosensitizer drugs based on compensatory stimulation of lactate release. Cancers. 2022;14:5454.
King MP, Attardi G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–3.
NavaneethaKrishnan S, Rosales J, Lee K-Y. ROS-mediated cancer cell killing through dietary phytochemicals. Oxid Med Cell Longev. 2019;2019:9051542.
Kim SJ, Kim HS, Seo YR. Understanding of ROS-inducing strategy in anticancer therapy. Oxid Med Cell Longev. 2019;2019:5381692.
Kovalski JR, Kuzuoglu-Ozturk D, Ruggero D. Protein synthesis control in cancer: selectivity andtherapeutic targeting. EMBO J. 2022;41: e109823.
Liu J, Xu Y, Stoleru D, Salic A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Nat Acad Sci USA. 2011;109:413–8.
Bedi M, Ray M, Ghosh A. Active mitochondrial respiration in cancer: a target for the drug. Mol Cell Biochem. 2022;477:345–61.
Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, Rohlena J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. BBA-Mol Basi Dis. 2020;1866: 165759.
Takahashi S, Shinomiya T, Nagahara Y. Azoxystrobin induces apoptosis and cell cycle arrest in human leukemia cells independent of p53 expression. Anticancer Res. 2022;42:1307–12.
Niego AG, Raspé O, Thongklang N, Charoensup R, Lumyong S, Stadler M, Hyde KD. Taxonomy, diversity and cultivation of the oudemansielloid/xeruloid taxa Hymenopellis , Mucidula , Oudemansiella , and Xerula with respect to their bioactivities: A review. J Fungi. 2021;7:51.
Li L, Li J, Chen H, Shen Y, Lu Y, Zhang M, Tang X. Azoxystrobin induces apoptosis via PI3K/AKT and MAPK signal pathways in oral leukoplakia progression. Front Pharmacol. 2022;13: 912084.
Forkink M, Basit F, Teixeira J, Swarts HG, Koopman WJH, Williems PHGM. Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol. 2015;6:607–16.
Download references
Acknowledgements
This work was supported by JSPS KAKENHI (Grant Number JP21KK0102). Mucidula venosolamellata (TUFC 30333) was provided by Fungus/Mushroom Resource and Research Center, Tottori University, with support in part from the National BioResource Project (NBRP), MEXT, Japan.
Author information
Authors and affiliations.
Department of Veterinary Pharmacology, Faculty of Agriculture, Tottori University, Tottori, 680-8553, Japan
Kenji Takahashi & Toshio Ohta
Division of Functional Fungal Physiology and Pharmacology, Fungus/Mushroom Resource and Research Center, Faculty of Agriculture, Tottori University, Tottori, 680-8553, Japan
Graduate School of Sustainability Sciences, Tottori University, Tottori, 680-8553, Japan
Tomoya Tanaka
Division of Applied Fungal Chemistry, Fungus/Mushroom Resource and Research Center, Faculty of Agriculture, Tottori University, Tottori, 680-8553, Japan
Atsushi Ishihara
You can also search for this author in PubMed Google Scholar
Contributions
KT designed and conducted the experiment and wrote the manuscript. AI and TT prepared the materials. TO supported the execution of research, and AI and TO wrote, reviewed, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Correspondence to Toshio Ohta .
Ethics declarations
Ethics approval and consent to participate, consent for publication, competing interests.
The authors declare that they have no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Takahashi, K., Tanaka, T., Ishihara, A. et al. Strobilurin X acts as an anticancer drug by inhibiting protein synthesis and suppressing mitochondrial respiratory chain activity. Discov Onc 15 , 177 (2024). https://doi.org/10.1007/s12672-024-01041-w
Download citation
Received : 07 November 2023
Accepted : 17 May 2024
Published : 20 May 2024
DOI : https://doi.org/10.1007/s12672-024-01041-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Mucidula venosolamellata
- Strobilurin X
- Mitochondrial respiratory chain complex III
- Protein synthesis
- Find a journal
- Publish with us
- Track your research
Chemical Communications
Stereoselective synthesis of α-glycosyl azides: allyl glycosyl sulfones as radical precursors.
Despite their critical importance in drug development and biochemistry, efficiently synthesizing α-glycosyl azides has continued to pose significant challenges. In this report, we introduce a universal and practical radical reaction for the stereoselective synthesis of α-glycosyl azides using bench-stable allyl glycosyl sulfones as the donor. This method is characterized by its mild reaction conditions, high stereoselectivity, and extensive scope of glycosyl units. Moreover, the accessibility of several structurally complex drug-sugar conjugates underscores the practicality of our approach.
Supplementary files
- Supplementary information PDF (5201K)
Article information
Download citation, permissions.
Y. Li, Y. Tian, D. Xie, Y. Wang and D. Niu, Chem. Commun. , 2024, Accepted Manuscript , DOI: 10.1039/D4CC01687D
To request permission to reproduce material from this article, please go to the Copyright Clearance Center request page .
If you are an author contributing to an RSC publication, you do not need to request permission provided correct acknowledgement is given.
If you are the author of this article, you do not need to request permission to reproduce figures and diagrams provided correct acknowledgement is given. If you want to reproduce the whole article in a third-party publication (excluding your thesis/dissertation for which permission is not required) please go to the Copyright Clearance Center request page .
Read more about how to correctly acknowledge RSC content .
Social activity
Search articles by author.
This article has not yet been cited.
Advertisements
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- My Account Login
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Open access
- Published: 18 May 2024
Design, synthesis, molecular docking study, and α-glucosidase inhibitory evaluation of novel hydrazide–hydrazone derivatives of 3,4-dihydroxyphenylacetic acid
- Hammad Khan 1 ,
- Faheem Jan 2 , 3 ,
- Abdul Shakoor 4 ,
- Ajmal Khan 5 ,
- Abdullah F. AlAsmari 6 ,
- Fawaz Alasmari 6 ,
- Saeed Ullah 5 ,
- Ahmed Al-Harrasi 5 ,
- Momin Khan 4 &
- Shaukat Ali 1
Scientific Reports volume 14 , Article number: 11410 ( 2024 ) Cite this article
384 Accesses
6 Altmetric
Metrics details
- Biochemistry
- Chemical biology
- Drug discovery
A series of novel Schiff base derivatives (1–28) of 3,4-dihydroxyphenylacetic acid were synthesized in a multi-step reaction. All the synthesized Schiff bases were obtained in high yields and their structures were determined by 1 HNMR, 13 CNMR, and HR-ESI–MS spectroscopy. Except for compounds 22 , 26 , 27, and 28, all derivatives show excellent to moderate α-glucosidase inhibition. Compounds 5 (IC 50 = 12.84 ± 0.52 µM), 4 (IC 50 = 13.64 ± 0.58 µM), 12 (IC 50 = 15.73 ± 0.71 µM), 13 (IC 50 = 16.62 ± 0.47 µM), 15 (IC 50 = 17.40 ± 0.74 µM), 3 (IC 50 = 18.45 ± 1.21 µM), 7 (IC 50 = 19.68 ± 0.82 µM), and 2 (IC 50 = 20.35 ± 1.27 µM) shows outstanding inhibition as compared to standard acarbose (IC 50 = 873.34 ± 1.67 µM). Furthermore, a docking study was performed to find out the interaction between the enzyme and the most active compounds. With this research work, 3,4-dihydroxyphenylacetic acid Schiff base derivatives have been introduced as a potential class of α-glucosidase inhibitors that have remained elusive till now.
Similar content being viewed by others

Aryl-quinoline-4-carbonyl hydrazone bearing different 2-methoxyphenoxyacetamides as potent α-glucosidase inhibitors; molecular dynamics, kinetic and structure–activity relationship studies

Novel N′-substituted benzylidene benzohydrazides linked to 1,2,3-triazoles: potent α-glucosidase inhibitors

Synthesis, in vitro inhibitor screening, structure–activity relationship, and molecular dynamic simulation studies of novel thioquinoline derivatives as potent α-glucosidase inhibitors
Introduction.
Diabetes mellitus is a persistent and potentially life-threatening metabolic condition characterized by inadequate insulin secretion, resulting in a complication known as hyperglycaemia 1 , 2 , 3 , 4 . In type II diabetes mellitus, linked to increased postprandial glucose levels, there is an elevated risk of stroke, atherosclerosis, and other cardiovascular conditions 5 , 6 . Therefore, an effective approach to managing type II diabetes mellitus and its associated issues involves the inhibition of digestive enzymes, specifically aimed at alleviating postprandial hyperglycemia 7 , 8 , 9 . As enzyme activity and blood glucose concentrations are strongly correlated with each other, the inhibition of α-glucosidase can potentially decrease postprandial blood glucose levels 10 , 11 . The function of α-glucosidase inhibitors like acarbose, miglitol, and voglibose, used in clinical settings is to slow down sharp rises in blood sugar levels 12 , 13 . The α-glucosidase enzyme found in human intestinal cells serves a crucial role as the primary hydrolase enzyme. Its primary function involves breaking down complex carbohydrates into glucose monomers, which can then readily diffuse into the bloodstream 14 , 15 . In the case of type II diabetes, where human body is resistant to insulin then α-glucosidase inhibitors utilization plays a beneficial role in reducing postprandial hyperglycaemia 16 , 17 . Commonly available α-glucosidase inhibitors such as acarbose, miglitol, and voglibose are associated with side effects such as diarrhea, nausea, and other intestinal disturbances 18 , 19 . Notably, numerous pieces of evidences indicate that α-glucosidase inhibitors may prove beneficial in the treatment of carbohydrate-mediated diseases, including conditions such as cancer, Alzheimer’s disease, hepatitis, and bacterial and viral infections 20 , 21 , 22 . Consequently use of α-glucosidase inhibitors has emerged as a promising therapeutic avenue for mitigating the risks associated with diabetes and related conditions 23 , 24 . Therefore, the pursuit of designing and developing novel α-glucosidase inhibitors with high efficacy and minimal side effects holds a great appeal for medicinal chemists.
Schiff bases are one of the most widely used organic compounds having a broad range of applications in different fields such as biology, medicinal drugs, organometallic chemistry, inorganic and analytical chemistry 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 . Imine or azomethine groups are mostly present in natural and-synthetic compounds 33 , 34 , 35 . The presence of imine or azomethine group in Schiff bases has shown profound biological importance and have been recognized as privileged precursors for designing biologically active drugs 36 , 37 , 38 with a broad spectrum of biological activities 39 . For instance compounds ( A ) and ( B ) show antibacterial activity 40 , 41 , 42 , compound ( C ) and ( D ) have anticancer activity 43 , 44 , compound ( E ) shows anti-cholinesterase activity 45 , compound ( F ) shows anti-inflammatory activity 46 , compound ( G ) shows anti-fungal activity 47 , compound ( H ) shows anti-viral activity 48 , and compound ( I ) shows anti-oxidant activity 49 ( Fig. 1 ) . Knowing the biological importance of Schiff bases, this study focuses on synthesizing novel Schiff base derivatives as potent inhibitors of α-glucosidase. These synthetic compounds are expected to possess high lipophilicity, enhanced activity, and facilitating easy passage through cellular barriers.

Commercially available drugs containing Schiff base moities.
Most of the synthetic anti-diabetic drugs are widely used now days because they are synthesized from cheaper reagent and are effective and safer than most of natural anti-diabetic drugs. On the basis of literature survey, the current research work is focused mainly on the synthesis of 3,4-dihydroxyphenylacetic acid derived Schiff bases, their molecular docking study and their α-glucosidase inhibition assay.
In the current work a series of 3,4-dihydroxyphenylacetic acid Schiff bases (1–28) were synthesized in good yields through multi-step reactions (Scheme 1 ). First 3,4-dihydroxyphenylacetic acid (a) was esterified to the corresponding esters (b) in ethanol and a small amount of sulfuric acid and then treated with excess amount of hydrazine hydrate to afford 3,4-dihydroxyphenylhydrazide (c) . Subsequently, Schiff bases of 3,4-dihydroxyphenylhydrazide were obtained by treating c with various aromatic aldehydes in ethanol and catalytic amount of glacial acetic acid. All the Schiff bases were recrystallized from ethanol in good yields (1–28) . The synthesized Schiff bases were characterized with the help of 1 H-NMR, 13 C-NMR, and HR-ESI–MS spectroscopy. Finally, Schiff bases of 3,4-dihydroxyphenylhydrazide were subjected to α-glucosidase inhibition assay.

Design for the synthesis of Schiff bases of 3,4-dihydroxyphenylhydrazide.
Materials and methods
3,4-Dihydroxyphenylacetic acid, hydrazine hydrate, ethanol, DMSO, potassium carbonate, and iodine were obtained from Macklin Company and used without further purification. Reactions were tracked using Merck aluminum TLC plates with 0.2 mm of silica gel 60 F-254. The 1 H-NMR and 13 C-NMR spectra of the synthesized compounds were recorded in DMSO-d 6 using an Avance Bruker AM 600 MHz spectrometer. Chemical shift values are expressed in ppm, and coupling constants (J) are reported in hertz (Hz). The molecular masses of the synthesized compounds were determined by high-resolution electrospray ionization mass spectrometry (HR-ESI–MS). Melting points of all the new synthesized compounds were determined using a Digital Electro-thermal apparatus.
Synthesis of ethyl 2-(2,4-dihydroxyphenyl)acetate (b)
In a 100 mL round-bottom flask, 3,4-dihydroxyphenylacetic acid ( a ) (1.68 g, 10 mmol) was dissolved in 35 mL of absolute ethanol. The temperature of this solution was lowered to 0 °C using an ethanol–water ice bath. Subsequently, 0.5 mL of concentrated sulfuric acid (H 2 SO 4 ) was added drop wise as a dehydrating agent, and the mixture was stirred at room temperature for 2 h before being refluxed for an additional 8 h. After cooling the reaction mixture to room temperature, it was poured onto a beaker containing 20 mL of crushed ice. The esterified product (monitored via TLC) was collected through filtration and neutralized with a 5% aqueous solution of sodium bicarbonate (NaHCO 3 ), yielding 1.79 g of ethyl 2-(3,4-dihydroxyphenyl)acetate ( b ).
Synthesis of 2-(3,4-dihydroxyphenyl)acetohydrazide (c)
Ethyl 2-(3,4-dihydroxyphenyl)acetate ( b ) (0.98 g, 5 mmol) was dissolved in 10 mL of absolute ethanol, and 15 mmol of 80% hydrazine hydrate was gradually added. The reaction mixture was refluxed for 8–10 h, monitored by TLC using a solvent system of n-hexane and ethyl acetate (1:2). Upon completion of the reaction, the mixture was cooled to room temperature, resulting in the appearance of a precipitate. The precipitate was filtered, washed with an excess of distilled water (100 mL), and then dried in an oven set at 50 °C, yielding 0.87 g of the desired product ( c ).
Synthesis of Schiff bases (1–28)
One millimole of 3,4-dihydroxyphenylacetic acid hydrazide was subjected to a reaction with an equimolar amount of aromatic aldehyde in the presence of 5 mL of absolute ethanol as a solvent and a few drops of glacial acetic acid as catalyst. The reaction mixture was refluxed and allowed to stir overnight, with monitoring through TLC (ethyl acetate: n-hexane: methanol 1:1:0.5). The resulting precipitate was cooled to room temperature, filtered, dried, and subsequent recrystallizion from ethanol afforded Schiff bases ( 1–28 ) . The structural characterization of the synthesized Schiff bases was conducted through 1 H-NMR, 13 C-NMR, and HR-ESI–MS spectroscopy.
Results and discussions
In vitro α-glucosidase inhibition effect.
All synthetic novel Schiff bases of 3,4-dihydroxypehnylacetic acid derivatives (1–28) were subjected to α-glucosidase inhibition effect according to the literature protocol 50 . Except compounds 22 , 26 , 27 , and 28 , all derivatives show good to moderate α-glucosidase inhibition potential ranging from (IC 50 = 12.84 ± 0.52–43.76 ± 2.34 µM) as compared to standard acarbose (IC 50 = 873.34 ± 1.67 µM). Compounds, 12 (IC 50 = 15.73 ± 0.71 µM), 15 (IC 50 = 17.40 ± 0.74 µM), 7 (IC 50 = 19.68 ± 0.82 µM), 4 (IC 50 = 13.64 ± 0.58 µM), 13 (IC 50 = 16.62 ± 0.47 µM), 2 (IC 50 = 20.35 ± 1.27 µM), 3 (IC 50 = 18.45 ± 1.21 µM) and 5 (IC 50 = 12.84 ± 0.52 µM) were found to be more potent among the series Table 1 .
Structure–activity relationship (SAR)
Structure activity relation for α-glucosidase inhibition potential was developed from Table 1 by analyzing the effect of various aldehyde substituents, R and data of inhibition of the most active compounds as shown in Fig. 2 . α-glucosidase inhibition activity of hydroxyl-substituted analogs ( 1–5 ) provides valuable insights into structure–activity relationships (SAR), with a focus on the positional effect of hydroxy groups. All derivatives showed excellent inhibition effect, among the analogs tested, the ortho-hydroxy analog 1 exhibited an excellent inhibition activity, with an IC 50 value of 24.37 ± 0.83, indicating a potential role for hydroxyl substitution at ortho position. Similarly, the para-hydroxy analog 2 demonstrated comparable activity, with an IC 50 value of 20.35 ± 1.27, suggesting that hydroxyl substitution at para position also contributes to inhibition potency. The introduction of a second hydroxyl group at positions meta and para position of compound 3 yielded a slightly increased inhibition activity, with an IC 50 value of 18.45 ± 1.21, highlighting the importance of the proximity and arrangement of hydroxyl groups. Further examination of another ortho, para dihydroxy analog 4 revealed a notable augmentation in inhibition activity, with an IC 50 value of 13.64 ± 0.58. Most notably, the ortho, meta, and para trihydroxy analog exhibited the highest inhibition activity among the hydroxyl substituted analogs tested, with an IC 50 value of 12.84 ± 0.52. However, it is crucial to note that all analogs displayed excellent inhibition activity compared to the standard inhibitor, acarbose, which had an IC 50 value of 873.34 ± 1.67.

Structure Activity relationship of compounds 2, 3, 4, 5, 7, 12, 13, and 15.
Compounds 6–10 represent analogs where the hydrogen of one hydroxyl group has been substituted with either methoxy (compounds 6 , 7 , and 8 ) or ethoxy (compounds 9 and 10 ). The IC 50 values for these analogs, namely 6 (IC 50 = 23.69 ± 1.11), 7 (IC 50 = 19.68 ± 0.82), 8 (IC 50 = 20.88 ± 0.53), 9 (IC 50 = 27.29 ± 1.15), and 10 (IC 50 = 28.66 ± 1.21), indicate that the introduction of these electron-donating groups results in a slight decrease in inhibition activity. Moreover, the impact of the ethoxy group appears to be more pronounced than that of the methoxy group, as evidenced by the observed results. Compound 11 , featuring a hydroxyl group at the meta position and a methyl group at the para position, exhibited an IC 50 value of 33.51 ± 1.22, indicating a decrease in activity when compared to compounds 1–10 .
Compound 12 , with an IC 50 of 15.73 ± 0.71, is ortho-methoxy substituted, while compound 13 , with an IC 50 of 16.62 ± 0.47, is para-methoxy substituted. Compound 14 , an analog with meta, para-dimethoxy substitution, exhibits an IC 50 of 20.61 ± 0.59. Additionally, Compound 15 , featuring a para N,N -dimethylamino substituent, displays an IC 50 of 17.405 ± 0.74, while compound 16 (IC 50 of 26.41 ± 1.10) and 17 (IC 50 of 43.76 ± 2.34) with a methyl group at the para position and naphthalene respectively. The activity trends observed among these analogs underscore the significant role that both the position and nature of the substituent play in modulating inhibition activity.
Analogs 18–22 represent chloro-substituted variations. Analog 18 (ortho-chloro) exhibits an IC 50 value of 28.16 ± 1.09, analog 19 (para-chloro) shows an IC 50 value of 31.22 ± 1.40, and analog 20 (meta-chloro) demonstrates an IC 50 value of 27.26 ± 0.83. These analogs, each singly substituted with a chloro group, display no significant variation in activity based on the position of the chloro group. However, compound 21, ortho meta dichloro-substituted, displays an enhanced activity with an IC 50 value of 24.16 ± 0.52, compared to its singly-substituted counterpart. Conversely, compound 22 ortho ortho dichloro-substituted, exhibits no inhibitory activity, indicating an extreme positional effect.
Analogs 23 (with an IC 50 value of 30.46 ± 1.28), 24 (with an IC 50 value of 29.67 ± 0.98), and 25 (with an IC 50 value of 34.46 ± 1.48) represent para, ortho, and meta nitro-substituted variations, respectively. The inhibitory results of these analogs also suggest that the impact of the nitro group's position is relatively minor. Conversely, compounds 26–28 exhibited less than 50% inhibition, rendering them inactive.
In summary, the fundamental structure within this class of compounds predominantly dictates their activity. However, the specific characteristics of substituents within the structure exert a notable influence; for instance, hydroxyl substitutions, as well as those with electron-donating or electron-withdrawing properties, can either enhance or diminish the inhibitory activity. Moreover, introducing disubstitutions tends to amplify the effect on inhibition activity. Nonetheless, the positional arrangement of substituents appears to exert a relatively minor influence on inhibition activity.
Molecular docking
The docking study was carried out using the AutoDock Vina package (1.5.7 version) 51 . The docking study provides deep insight into synthesized compounds’ binding modes with α-glucosidase. The protein structure of α-glucosidase was obtained from Protein Data Bank [ https://www.rcsb.org/structure/3WY1 ]. The water molecules were eliminated from the protein structure, and the missing hydrogens were added to the protein structure using AutoDock Tools. For the docking calculations, a cube grid box was prepared with dimensions of 3.391, 1.310, and − 8.362 Å in x, y, and z -directions. Furthermore, the grid box position was centered on the middle of compounds under docking. The AutoGrid Tools was used to form grid maps, and spacing within the grid was selected as 0.375 Å. The grid consists of 40 points along x, y, and z -directions. We focused on the amino acids that play a key role in the binding with the active site of targeted compounds, as we listed in Table 2 .
The ligands (compounds) were made and optimized using the density functional theory (DFT) method. After the optimization, the ligands were used for docking calculation. The interactions between the ligands and amino acids of protein were visualized by the PyMOL 2.5.4. The visualization clearly shows the different types of interaction, such as van der Waals, attractive charges, conventional H-bonding, unfavorable interaction, and different types of π-interactions as shown in Fig. 3 . The Biovia Discovery Studio was used for making the 2D representation of the interaction between protein and ligand.

Docking studies of synthesized compounds 12, 15, 13, and 2 , respectively.
The molecular docking analysis reveals complex interactions between compound 12 and various amino acid residues. In particular, THR.445, LYS.352, and ASN.46 engage in conventional hydrogen bonding with the oxo groups of the compound. On the other hand, ALA.45, ALA.451, LEU.433, ASN.447, and SER.44 form van der Waals interactions with the ligand. Additionally, HIS.348, ASP.441, and ARG.450 exhibit π-interactions with the ring's delocalized electronic cloud, as shown in Table 2 and Fig. 3 . Furthermore, ALA.444 reveals π-sigma bonding with the aromatic ring. Notably, HIS.348 and ASN.443 contribute to unfavorable interactions with the ligand's oxo groups. The overall binding affinity of compound 12 is calculated as − 7.5 kcal/mol.
Compound 15 , shows a higher binding affinity of − 8.0 kcal/mol. This higher affinity reflects a substantial increase in the number of amino acid residues engaged in the interaction. Noteworthy contributors include LEU.446, LEU.433, LYS.352, ALA.514, and ASN.443, indicating their significant involvement through van der Waals interactions. Key amino acids, namely THR.445, LYS.352, and ASN.46, establish conventional hydrogen bonds with the OH and NH groups. The HIS.348 and ASP.441 participate in π-electronic interactions with the aromatic ring and NH through attractive charges/π-cation interactions, as shown in Fig. 3 . Furthermore, ALA.444, ALA.451, and ALA.454 contribute through π-alkyl interactions.
Compound 13 exhibits the highest binding affinity at − 8.2 kcal/mol. Notably, conventional hydrogen bonding is reduced, with only ASP.441 forming such bonds with the NH group of synthesized compound. In contrast, van der Waals interactions significantly increase compared to other compounds, with noteworthy contributions from amino acids such as VAL.513, ALA.514, GLU.432, LEU.433, and others, as listed in Table 2 . Furthermore, the ARG.450, ARG.437, ALA.451, and ALA.454 engage in π-interactions with the aromatic ring. These interactions highlight the unique binding characteristics of Compound 13 .
Compound 2 shows the binding affinity as − 7.9 kcal/mol. Notably, in Compound 2 , an increased number of amino acids, including HIS.348, ARG.437, ASN.447, ASP.441, and ALA.451, actively contribute through conventional H-bonding. The engagement of oxygen and nitrogen atoms from compound 2 in H-bonding is shown in Fig. 3 . The multifaceted role of HIS.348 not only participates in conventional hydrogen bonding but also engages in π-cation interactions with the aromatic ring. Furthermore, Compound 2 is surrounded by many amino acids through van der Waals interactions, as detailed in Table 2 . Additionally, ARG.450 and ALA.454 contribute through π-interactions. After an in-depth molecular docking analysis, compounds 12, 15, 13, and 2 exhibited diverse interaction profiles with the target protein. These findings underscore their unique binding characteristics, providing valuable insights for their potential development as promising drug discovery and optimization candidates.
A series of novel Schiff base derivatives ( 1–28 ) of 3,4-dihydroxyphenylacetic acid were successfully synthesized and characterized using advanced spectroscopic techniques, including 1 H-NMR, 13 C-NMR, and high-resolution electrospray ionization mass spectrometry (HR-ESI–MS). Subsequently, the inhibitory potential of these compounds against the α-glucosidase enzyme was evaluated. Most of our synthesized compounds exhibited satisfactory to moderate inhibitory activity against α-glucosidase (see Supplementary information ). Notably, derivatives 12 , 15 , 7 , 4 , 13 , 2 , 3 , and 5 demonstrated outstanding inhibition, showcasing IC 50 values ranging between 12.84 ± 0.52 µM and 20.35 ± 1.27 µM. To further elucidate the binding modes of these promising derivatives with the enzyme active site, molecular docking studies were conducted. This comprehensive approach provides valuable insights into the synthesized compounds' inhibitory potential and their potential application in developing α-glucosidase inhibitors.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
Khunti, K. et al. Diabetes and multiple long-term conditions: A review of our current global health challenge. Diabetes Care 46 , 2092–2101 (2023).
Article PubMed Google Scholar
Durmaz, L. et al. Screening of carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase, and α-glycosidase enzyme inhibition effects and antioxidant activity of coumestrol. Molecules 27 , 3091 (2022).
Article CAS PubMed PubMed Central Google Scholar
Cakmak, K. C. & Gülçin, İ. Anticholinergic and antioxidant activities of usnic acid-an activity-structure insight. Toxicol. Rep. 6 , 1273–1280 (2019).
Article Google Scholar
Durmaz, L. et al. Antioxidant, antidiabetic, anticholinergic, and antiglaucoma effects of magnofluorine. Molecules 27 , 5902 (2022).
Matheus, A. S. M. et al. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013 , 1–15. https://doi.org/10.1155/2013/653789 (2013).
Article CAS Google Scholar
Yi, J. et al. Polypeptide from moschus suppresses lipopolysaccharide-induced inflammation by inhibiting NF-κ B-ROS/NLRP3 pathway. Chin. J. Integr. Med. 29 , 895–904 (2023).
Article CAS PubMed Google Scholar
Toeller, M. α-Glucosidase inhibitors in diabetes: efficacy in NIDDM subjects. Eur. J. Clin. Investig. 24 , 31–35 (1994).
Cui, G. et al. Synthesis and characterization of Eu (III) complexes of modified cellulose and poly (N-isopropylacrylamide). Carbohydr. Polym. 94 , 77–81 (2013).
Derosa, G. & Maffioli, P. α-Glucosidase inhibitors and their use in clinical practice. Arch. Med. Sci. AMS 8 , 899 (2012).
Wang, Y., Zhai, W., Cheng, S., Li, J. & Zhang, H. Surface-functionalized design of blood-contacting biomaterials for preventing coagulation and promoting hemostasis. Friction 11 , 1371–1394 (2023).
Zhang, B.-W. et al. Dietary flavonoids and acarbose synergistically inhibit α-glucosidase and lower postprandial blood glucose. J. Agric. Food Chem. 65 , 8319–8330 (2017).
Smith, D. L., Orlandella, R. M., Allison, D. B. & Norian, L. A. Diabetes medications as potential calorie restriction mimetics—a focus on the alpha-glucosidase inhibitor acarbose. GeroScience 43 , 1123–1133 (2021).
Wang, W. et al. Macrophage-derived biomimetic nanoparticles for light-driven theranostics toward Mpox. Matter 7 , 1187–1206 (2024).
Lin, A.H.-M., Lee, B.-H. & Chang, W.-J. Small intestine mucosal α-glucosidase: A missing feature of in vitro starch digestibility. Food Hydrocoll. 53 , 163–171 (2016).
Cui, G. et al. Synthesis and characterization of phenylboronic acid-containing polymer for glucose-triggered drug delivery. Sci. Technol. Adv. Mater. 21 , 1–10 (2020).
Bai, R. et al. Second generation β-elemene nitric oxide derivatives with reasonable linkers: Potential hybrids against malignant brain glioma. J. Enzyme Inhib. Med. Chem. 37 , 379–385 (2022).
Joshi, S. R. et al. Therapeutic potential of α-glucosidase inhibitors in type 2 diabetes mellitus: An evidence-based review. Expert Opin. Pharmacother. 16 , 1959–1981 (2015).
Leroux‐Stewart, J., Rabasa‐Lhoret, R. & Chiasson, J. L. α‐Glucosidase inhibitors. In International Textbook of Diabetes Mellitus , 673–685 (2015).
Zhang, Y. et al. Ephedra Herb extract ameliorates adriamycin-induced nephrotic syndrome in rats via the CAMKK2/AMPK/mTOR signaling pathway. Chin. J. Nat. Med. 21 , 371–382 (2023).
CAS PubMed Google Scholar
Khan, M. et al. Synthesis, molecular modeling and biological evaluation of 5-arylidene-N, N-diethylthiobarbiturates as potential α-glucosidase inhibitors. Med. Chem. 15 , 175–185 (2019).
Zhao, J. et al. Tumor cell membrane-coated continuous electrochemical sensor for GLUT1 inhibitor screening. J. Pharm. Anal. 13 , 673–682 (2023).
Article PubMed PubMed Central Google Scholar
Zhang, Z., Zhang, W., Hou, Z.-W., Li, P. & Wang, L. Electrophilic halospirocyclization of N-benzylacrylamides to access 4-halomethyl-2-azaspiro [4.5] decanes. J. Org. Chem. 88 , 13610–13621 (2023).
Gul, S. et al. Synthesis, molecular docking and DFT analysis of novel bis-Schiff base derivatives with thiobarbituric acid for α-glucosidase inhibition assessment. Sci. Rep. 14 , 3419 (2024).
Cui, G. et al. Synthesis and characterization of Eu (III) complexes of modified D-glucosamine and poly (N-isopropylacrylamide). Mater. Sci. Eng. C 78 , 603–608 (2017).
Jia, Y. & Li, J. Molecular assembly of Schiff base interactions: Construction and application. Chem. Rev. 115 , 1597–1621 (2015).
Jamil, W. et al. Phenoxyacetohydrazide Schiff bases: β-Glucuronidase inhibitors. Molecules 19 , 8788–8802 (2014).
Chen, H. & Rhodes, J. Schiff base forming drugs: Mechanisms of immune potentiation and therapeutic potential. J. Mol. Med. 74 , 497–504 (1996).
Aziz, A. N. et al. Synthesis, crystal structure, DFT studies and evaluation of the antioxidant activity of 3, 4-dimethoxybenzenamine Schiff bases. Molecules 19 , 8414–8433 (2014).
Jamil, W. et al. Synthesis, anti-diabetic and in silico QSAR analysis of flavone hydrazide Schiff base derivatives. J. Biomol. Struct. Dyn. 40 , 12723–12738 (2022).
Yiğit, B., Yiğit, M., Taslimi, P., Gök, Y. & Gülçin, İ. Schiff bases and their amines: Synthesis and discovery of carbonic anhydrase and acetylcholinesterase enzymes inhibitors. Archiv Pharm. 351 , 1800146 (2018).
Buldurun, K. et al. Synthesis, characterization, powder X-ray diffraction analysis, thermal stability, antioxidant properties and enzyme inhibitions of M (II)-Schiff base ligand complexes. J. Biomol. Struct. Dyn. 39 , 6480–6487 (2021).
Aytac, S., Gundogdu, O., Bingol, Z. & Gulcin, İ. Synthesis of Schiff bases containing phenol rings and investigation of their antioxidant capacity, anticholinesterase, butyrylcholinesterase, and carbonic anhydrase inhibition properties. Pharmaceutics 15 , 779 (2023).
Da Silva, C. M. et al. Schiff bases: A short review of their antimicrobial activities. J. Adv. Res. 2 , 1–8 (2011).
Article ADS Google Scholar
Sykula, A. D. A. Schiff bases as important class of pharmacological agents. J. Pharm. Pharmacol. 6 , 989–1009 (2018).
Google Scholar
Khan, M. et al. Synthesis of new bis (dimethylamino) benzophenone hydrazone for diabetic management: In-vitro and in-silico approach. Heliyon 10 , e23323 (2024).
Sahu, R. & Thakur, D. Schiff base: An overview of its medicinal chemistry potential for new drug molecules. Int. J. Pharm. Sci. Nanotechnol. (IJPSN) 5 , 1757–1764 (2012).
Xu, W.-X. et al. Personalized application of antimicrobial drugs in pediatric patients with augmented renal clearance: a review of literature. European Journal of Pediatrics , 1–10 (2023).
Riaz, M. et al. Unraveling the molecular structure, spectroscopic properties, and antioxidant activities of new 2, 4-dinitrophenylhydrazone derivatives through a comprehensive investigation. Arabian Journal of Chemistry 16 , 105259 (2023).
Khan, M. et al. Green synthesis, characterization, DPPH and ferrous Ion-chelating (FIC) activity of Tetrakis-Schiff’s bases of terephthalaldehyde. Lett. Drug Design Discov. 16 , 249–255 (2019).
Durgun, M. et al. Synthesis, characterisation, biological evaluation and in silico studies of sulphonamide Schiff bases. J. Enzyme Inhib. Med. Chem. 35 , 950–962 (2020).
Ashraf, M. A., Mahmood, K., Wajid, A., Maah, M. J. & Yusoff, I. Synthesis, characterization and biological activity of Schiff bases. IPCBEE 10 , 185 (2011).
Xavier, A. & Srividhya, N. Synthesis and study of Schiff base ligands. IOSR J. Appl. Chem. 7 , 06–15 (2014).
Sadia, M. et al. Schiff base ligand L synthesis and its evaluation as anticancer and antidepressant agent. J. King Saud Univ. Sci. 33 , 101331 (2021).
Suyambulingam, J. K. et al. Synthesis, structure, biological/chemosensor evaluation and molecular docking studies of aminobenzothiazole Schiff bases. J. Adhes. Sci. Technol. 34 , 2590–2612 (2020).
Raza, R. et al. Synthesis and biological evaluation of 3-thiazolocoumarinyl schiff-base derivatives as cholinesterase inhibitors. Chem. Biol. Drug Design 80 , 605–615 (2012).
Liaras, K., Fesatidou, M. & Geronikaki, A. Thiazoles and thiazolidinones as COX/LOX inhibitors. Molecules 23 , 685 (2018).
El-Tabl, A. S., Shakdofa, M. M., El-Seidy, A. M. & Al-Hakimi, A. N. Synthesis, characterization and antifungal activity of metal complexes of 2-(5-((2-chlorophenyl) diazenyl)-2-hydroxybenzylidene) hydrazinecarbothioamide. Phosphorus Sulfur Silicon Relat. Elem. 187 , 1312–1323 (2012).
Kumar, K. S., Ganguly, S., Veerasamy, R. & De Clercq, E. Synthesis, antiviral activity and cytotoxicity evaluation of Schiff bases of some 2-phenyl quinazoline-4 (3) H-ones. Eur. J. Med. Chem. 45 , 5474–5479 (2010).
Mermer, A. et al. Synthesis of novel Schiff bases using green chemistry techniques; antimicrobial, antioxidant, antiurease activity screening and molecular docking studies. J. Mol. Struct. 1181 , 412–422 (2019).
Article ADS CAS Google Scholar
Pierre, C., Roland, R., Tremblay, R. & Dube, J. p-Nitrophenol-α-D-glucopyranoside as substrate for measurement of maltase activity in human semen. Clin. Chem. 24 , 208–211 (1978).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 , 455–461 (2010).
Download references
Acknowledgements
Authors are thankful to researchers supporting project number (RSP2024R235), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and affiliations.
Organic Synthesis and Catalysis Research Laboratory, Institute of Chemical Sciences, University of Peshawar, Peshawar, 25120, Khyber Pakhtunkhwa, Pakistan
Hammad Khan & Shaukat Ali
Shenyang National Laboratory for Materials Science, Institute of Metal Research Chinese Academy of Sciences, Shenyang, 110016, Liaoning, China
School of Materials Science and Engineering, University of Science and Technology of China, Shenyang, 110016, Liaoning, China
Department of Chemistry, Abdul Wali Khan University, Mardan, 23200, Pakistan
Abdul Shakoor & Momin Khan
Natural and Medical Sciences Research Center, University of Nizwa, PO Box 33, 616, Birkat Al Mauz, Nizwa, Oman
Ajmal Khan, Saeed Ullah & Ahmed Al-Harrasi
Department of Pharmacology and Toxicology, College of Pharmacy, King Saud University, Riyadh, 11451, Saudi Arabia
Abdullah F. AlAsmari & Fawaz Alasmari
You can also search for this author in PubMed Google Scholar
Contributions
H.K. synthesized the compounds, while H.K., A.A., F.A., and M.K. performed structural elucidation and wrote the original draft of the manuscript. A.K., S.U., and A.A performed the α-glucosidase inhibitory activity of the compounds. F.J. and A.S. performed molecular docking study of the synthesized compounds. M.K. and S.A. designed and supervised the project, assisted in the writing, the reviewing, and the editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Correspondence to Ahmed Al-Harrasi , Momin Khan or Shaukat Ali .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary information., rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Khan, H., Jan, F., Shakoor, A. et al. Design, synthesis, molecular docking study, and α-glucosidase inhibitory evaluation of novel hydrazide–hydrazone derivatives of 3,4-dihydroxyphenylacetic acid. Sci Rep 14 , 11410 (2024). https://doi.org/10.1038/s41598-024-62034-x
Download citation
Received : 24 December 2023
Accepted : 13 May 2024
Published : 18 May 2024
DOI : https://doi.org/10.1038/s41598-024-62034-x
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
By submitting a comment you agree to abide by our Terms and Community Guidelines . If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.


An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- HHS Author Manuscripts
The DARK Side of Total Synthesis: Strategies and Tactics in Psychoactive Drug Production
Schuyler a. chambers.
† Department of Chemistry, Vanderbilt University, 7330 Stevenson Center, Nashville, Tennessee 37235, United States
Jenna M. DeSousa
Eric d. huseman, steven d. townsend.
‡ Institute of Chemical Biology, Vanderbilt University, 896 Preston Research Building, Nashville, Tennessee 37232, United States
Author Contributions
Humankind has used and abused psychoactive drugs for millennia. Formally, a psychoactive drug is any agent that alters cognition and mood. The term “psychotropic drug” is neutral and describes the entire class of substrates, licit and illicit, of interest to governmental drug policy. While these drugs are prescribed for issues ranging from pain management to anxiety, they are also used recreationally. In fact, the current opioid epidemic is the deadliest drug crisis in American history. While the topic is highly politicized with racial, gender, and socioeconomic elements, there is no denying the toll drug mis- and overuse is taking on this country. Overdose, fueled by opioids, is the leading cause of death for Americans under 50 years of age, killing ca. 64,000 people in 2016. From a chemistry standpoint, the question is in what ways, if any, did organic chemists contribute to this problem? In this targeted review, we provide brief historical accounts of the main classes of psychoactive drugs and discuss several foundational total syntheses that ultimately provide the groundwork for producing these molecules in academic, industrial, and clandestine settings.
Graphical abstract

1. INTRODUCTION
Psychoactive drugs affect cognition by influencing the synaptic operation of neurotransmitters in the central nervous system (CNS). 1 – 4 This action occurs through one of three general mechanisms: 5 (1) as agonists that mimic the operation of a neurotransmitter; (2) as antagonists that block the action of a neurotransmitter; or (3) by blocking the reuptake of neuro-transmitters at the synapse. In this regard, psychoactive drugs mimic or enhance naturally occurring states of consciousness; for example, sleeping pills promote drowsiness while benzodiazepines create a state of relaxation. Long-term use, whether medicinally or recreationally, often leads to tolerance, requiring the user to increase a drug’s dosage in order to achieve the baseline effect. As drug use increases, the user may also develop dependence in one of two forms. The first is psychological dependence, in which the user feels the urge to use the drug but abstinence poses no risk. The second form is physical dependence where serious physical and mental effects develop when a drug is withdrawn. In the United States millions of people use psychoactive drugs everyday ( Table 1 ). 6 While misuse is prominent, approximately one in three Americans are prescribed an opiate/opioid for pain management every year, 7 a testament to the importance of licit drug use for maintaining human health and wellness.
Psychoactive Drug Addiction in America a
2. SCOPE OF THIS REVIEW
2.1. the united states overdose epidemic..
While the goal of this survey is to review synthetic strategies used to produce the carbon frameworks of psychoactive drugs, it is undeniable that the scholarly exercises completed by organic chemists have guided illicit drug production. Thus, we would be neglectful to not briefly discuss the “DARK” and highly politicized nature of the topic. Virtually all known civilizations have used psychoactive drugs in an effort to self-medicate, anesthetize from emotional or physical trauma, or for recreational and religious purposes. In the United States, aggressive marketing of painkillers has led to a generation of individuals with opioid use disorders and a death toll closing in on 200 people per day. In both the second world war and Vietnam war, the Department of Defense employed “speed” or amphetamine to empower soldiers on the frontlines and sedatives and neuroleptics to prevent mental breakdowns from combat stress. 8 The crack-cocaine epidemic of the 1980s and early 1990s wreaked havoc among low-income, urban Americans. Similarly, “backyard”-produced methamphetamine reached its pinnacle among poorer rural Americans in the 1990s and early 21st century. One difference between those crises and the current iteration of the American drug epidemic is that opioid use disorder has its genesis at the pharmacy counter and crosses broad demographic groups.
2.2. Chemistry’s Role in the Crisis.
From a chemistry perspective, the current epidemic can likely be traced back to the introduction of OxyContin. “Oxy” is a semisynthetic opioid that shares structural homology with morphine, vide inf ra . Strong analgesics of this type have historically been reserved to manage pain associated with cancer therapy or surgery. During the mid-1990s oxycodone was marketed to treat chronic pain. As the use of opioids increased, pill distribution spread from primary-care practitioners to neighborhood dealers. Pills were also sold in large quantities from poorly regulated pain treatment centers colloquially known as “pill mills”. The problem appeared so rapidly that the American Public Health Association published a report in 2009 describing the prescription drug crisis titled “The promotion and marketing of OxyContin: Commercial triumph, public health tragedy”. 9 Ultimately, the closing of pill mills combined with an inability to treat addiction initiated a dark chapter in psychoactive drug use: the resurgence of heroin and the emergence of fentanyl. This has resulted in nearly 1000 deaths per week due to drug overdose, two-thirds of which are opioid related fatalities. The problem is so great that the illicit drug trade is currently the second most lucrative industry in the world, after weapons. 10
While the most problematic players in the drug crisis, and the largest section of this review, are opium alkaloids, they are not the only burdensome psychoactive drugs. In fact, deciding which agents are defined as drugs, how drug supply and use is regulated, and how society responds to drug users are hotly debated issues. The fact that the two drugs that cause the most impairment and damage in the world tobacco and alcohol are both legal in most countries illustrates this point. As does the fact that caffeine, the world’s most popular stimulant, is legal and unregulated worldwide. What role does organic chemistry play in the field of psychoactive drug synthesis and use? As one reads through the annals of published efforts, it is clear that legendary figures in the field, ranging from Larry Overman 11 and Phil Magnus, 12 to Kathy Parker 13 and James White, 14 have trained scores of students using these molecules as training tools. While the approaches are scholarly in nature, one cannot help but recognize the dichotomy: academic lessons assist in fueling both licit and clandestine drug use.
Given the level of intrigue that accompanies this class of molecules, several comprehensive accounts have been written to describe both the synthesis and the reactivity of these substrates. 15 – 18 The gap that this review fills is to compare and contrast syntheses in academic, industrial, and clandestine settings. Historically, academic pursuits have focused on demonstrating novel bond forming strategies and assembly tactics whereas industry has viewed their efforts as exercises in scale and efficiency. Meanwhile, the underworld of synthesis relies upon operational simplicity and material availability to make their syntheses practical. As a result, the de novo total syntheses of the academic world are commonly translated into large-scale semisyntheses in industrial settings that are further simplified for illicit manufacturing in makeshift clandestine laboratories. With this in mind, we chose to take a similar approach in the construction of this review and have highlighted various academic and industrial syntheses of the chosen psychoactive compounds before delving into their illicit synthesis as appropriate. It is our hope that this review will provide the reader with a foundation for delving deeper into the psychoactive drug literature and give context to how scholarly exercises are translated to clandestine drug synthesis.
3. THE OPIATES
3.1. historical perspective..
Opium extracts have been used for millennia. The earliest reference to opium production, in which the inhabitants of southern Mesopotamia cultivated Papaver somniferum , the opium poppy, predates 4000 B.C. 19 Known as Hul Gil, or the joy plant, the art of poppy culling was passed from the Sumerians to the Assyrians, Babylonians, and the ancient Egyptians. Eventually, its cultivation spread along the Silk Road from the Mediterranean to the Far East where the British sale of opium in China was the catalyst for the Opium Wars of the mid-1800s. 20 By way of definition, opiates are defined as any compounds, natural or semisynthetic, that share structural homology with opium alkaloids. We use the term opioid to refer to substances that elicit similar CNS effects but do not share structural homology with naturally occurring poppy alkaloids. 21 Regardless of their structural classification, opiates and opioids are large components of the illicit drug market, generating revenue in excess of $55 billion per year. 7 While the pharmacology of these molecules will be covered in detail in other parts of the DARK special issue, we briefly mention here that opiates and opioids act on the CNS to produce effects ranging from analgesia to sedation.
3.2. Structure.
The opiates ( Figure 1 ) mimic the activity of the endorphins, the body’s natural pain relievers. Structurally, the opiates feature a pentacyclic scaffold containing a phenolic A ring, a bridging bicyclic piperidine D ring, and a dihydrofuran like E ring ( Figure 1C ). While masked, most receptor active opiates and opioids share structural homology. These similarities are known as the “morphine rule” in which compounds generally incorporate into their carbon framework (1) a tertiary nitrogen with a small alkyl substituent; (2) a quaternary carbon; (3) a phenyl ring or its equivalent attached to the quaternary carbon; and (4) an ethyl linker between the quaternary carbon and the tertiary nitrogen. This complex architecture has served as the motivation for synthetic efforts spanning Gates’ synthesis of morphine in 1952 to the work of Fukuyama in 2017, vide infra .

Structures of various natural and semisynthetic opioid alkaloids. a Hydrocodone is also referred to as dihydrocodeinone. b Vicodin is a combination of hydrocodone and acetaminophen. c Percocet is a combination of oxycodone and acetaminophen. d Suboxone is a combination of naloxone and buprenorphine. Structural requirements of the so-called “morphine rule” are highlighted in pink.
3.3. Natural Opiates.
By weight the opium poppy P. somniferum contains ca. 8−19% morphine (1), 1−5% codeine (2), and 1−5% thebaine (4). Morphine and codeine are pharmaceutical agents, with morphine serving as an analgesic and codeine as a cough suppressant. Contrarily, oripavine (3) and thebaine are starting materials for semisynthetic opiates.
3.3.1. Morphine.
First isolated by Sertürner in the early 1800s, 22 morphine (1) was widely prescribed because it provides greater pain relief capacity than processed opium. Morphine is one of the most effective analgesics and is the standard against which new pain relievers are measured. Though the molecule’s structure, first proposed by Robinson 23 and later confirmed by Gates (total synthesis) 24 and Hodgkin (X-ray crystallography), 25 has inspired several syntheses, there is currently no practical source of morphine, either by chemical synthesis or through fermentation, that can compete with isolation. Nevertheless, synthetic efforts toward morphine have pushed the field of organic synthesis forward and have been appropriately recounted. 26 – 28 Consequently, this review would be remiss to attempt a full recapitulation of morphine’s rich synthetic history; instead, we aim to paint a broad picture and would direct the reader to the literature for a more detailed exhibition.
The review begins with the Gates’ total synthesis of morphine ( Scheme 1a ). 24 , 29 – 32 Starting from dienophile 14 , cycloaddition with butadiene provided racemic Diels−Alder adduct (±)- 15 in moderate yield, best represented as its enol tautomer (±)- 16 . 29 Cu 2 Cr 2 O 5 mediated reductive hydrogenation was accompanied by cyclization to give (±)- 17 , closing what would ultimately emerge as the piperidine D ring. Six further transformations, including a chiral resolution with L- (+)-dibenzoyl tartaric acid, gave enantiopure tetracycle (−)- 18 . At this junction, two major obstacles remained: (1) inversion of the C-14 stereocenter and (2) closure of the dihydrofuran-like ring. Thus, treatment of (−)- 18 with two equivalents of Br 2 followed by the addition of 2,4-dinitrophenyl hydrazine (NH 2 NHAr) produced an α,β -unsaturated hydrazone which underwent epimerization at C-14 to give the thermodynamically favored β -hydrogen substituted product. Hydrolysis of the hydrazone and chemoselective catalytic hydrogenation gave (−)- 19 which was exposed to excess Br 2 followed by 2,4-dinitrophenyl hydrazine to simultaneously close the E ring and install the requisite C7−C8 unsaturation. Hydrolysis of the hydrazone gave enone (−)- 20 which was converted to (−)-codeine (2) after reduction of the enone (in a 1,2 fashion) and aryl bromide. Finally, exposure to BBr 3 under modified Rapoport conditions gave (−)-morphine (1).

Three Total Syntheses of (−)-Morphine by the Gates, Gaunt, and Fukuyama Labs
In 2014, the Gaunt lab completed a formal synthesis of (−)-morphine ( Scheme 1b ). 33 The effort began with a seven step sequence highlighted by a Noyori transfer hydrogenation to give chiral aldehyde (−)-22 from isovanillin (21). A PhI(OAc) 2 mediated, intramolecular oxidative coupling was followed by Michael addition to give enone (+)-24. Next, the enone was elaborated to Boc-protected amine (−)-25. Luche reduction gave the corresponding allylic alcohol, which was heated under microwave irradiation with HCl to fragment the cyclic enol ether and deprotect the amine. Dehydrative ring closure, reductive amination, and carbamate protection gave (−)-28, completing the formal synthesis of (−)-morphine ( 1 ).
A third, noteworthy synthesis of morphine was reported by the Fukuyama lab in 2017 ( Scheme 1c ). 34 The synthesis began with the transformation of 7-methoxy-2-tetralone into chiral bicycle (+)- 29 using the d’Angelo approach. 35 Elimination of the tertiary alcohol accompanied acid mediated ring closure. Next, hydroselenation of the resulting olefin and oxidative elimination of the selenoxide gave vinyl tertiary alcohol (+)-30. A retro-aldol/aldol sequence provided tetracycle 31 which was transformed into diene (−)- 32 in five further steps. At this point Diels−Alder cycloaddition with singlet oxygen gave endoperoxide 33 which then underwent selective fragmentation to enone (+)- 34 . Elimination of the tertiary alcohol was followed by amine liberation and subsequent 1,6-conjugate addition to give a 1:1.4 ratio of neopinone to codeinone which converged to pure codeinone after acid treatment. Reduction of the enone gave (−)-codeine (2) and finally (−)-morphine (1) after treatment with BBr 3 .
3.3.2. Codeine.
Codeine ( 2 ) is prescribed for the relief of moderate pain and cough suppression. It has less analgesic ability than morphine and is usually taken orally. The recommended dose is 15−60 mg every 4−6 h, not exceeding 360 mg/day. Given its availability, codeine is commonly abused. When cough syrup is mixed with soft drinks, the brews are referred to as “lean” and “sizzurp”, among other names. Originating in Houston, Texas, lean gained popularity within southern hip-hop culture and has been associated with a number of deaths. Users risk overdose once consumption approaches 500 mg/day. Synthetically, codeine serves as a key intermediate in most morphine syntheses.
Our summary begins with Trost’s 2002 synthesis of (−)-codeine ( Scheme 2a ). 36 Corey−Fuchs olefination of chiral cyanoaldehyde (−)- 35 37 and regioselective dehalogenation gave (Z)-alkene (−)- 36 . Next, an intramolecular Heck reaction was used to close the B ring. SeO 2 oxidation of the resulting tetracycle (+)- 37 occurred preferentially at C 6 to give a mixture of alcohol and ketone (−)- 38 oxidation products that converged after a Dess−Martin periodinane oxidation. Next, the cyanoketone was reduced to the corresponding hydroimine. Subsequent amine formation occurred in the presence of methyl amine and reduction then gave (−)-39. Irradiation of precursor (−)-39 and LDA in THF afforded (−)-codeine (2) by way of an intramolecular hydroamination. Irradiation was critical as treatment with LDA alone did not result in cyclization.

Synthesis of Codeine by the Trost and Stork Labs
The Stork lab published a synthesis of (±)-codeine using a Diels−Alder cycloaddition to establish the B and C rings in a single move ( Scheme 2b ). 38 The synthesis began by exposing ( E )-1-methoxybut-1-en-3-yne to Schwartz’s reagent followed by the addition of aldehyde 41 to give (±)- 42 . Heating this compound in a sealed tube in the presence of Et 3 N produced the Diels−Alder adduct (±)- 43 as a 4:1 mixture of diastereomers. After 10 steps, amine (±)- 44 underwent 6-exo-tet closure to give (±)- 45 . Protection of the amine was followed by oxidation and LiAlH 4 reduction to give (±)-codeine ( 2 ).
3.4. Licit Semisynthetic Opiates.
While codeine and morphine are important analgesics, a number of semisynthetic opiates are also important therapeutics.
3.4.1. Hydromorphone.
(−)-Hydromorphone (5, Dilaudid) is a potent semisynthetic opiate. Structurally, it is identical to morphine with additional oxygenation at the C 6 position. As a result, it is more water-soluble than morphine and displays increased efficacy ( Table 2 ). 39
Opioid Conversion Chart a
Recently, the Hudlicky group published a second generation, chemoenzymatic formal synthesis of ent-hydromorphone ( Scheme 3 ). 40 Beginning with (2-bromoethyl)benzene ( 46 ), enzymatic dihydroxylation provided chiral diene (+)- 47 which could be elaborated to phenol (+)- 48 in seven additional steps. In a series of key transformations, oxidative phenol dearomatization was followed by Diels−Alder cycloaddition to establish the B and E rings in a single step. While Hudlicky’s first generation synthesis used a Pb(OAc) 4 oxidation and subsequent heating to give the desired Diels−Alder adduct in 50% yield, 41 the second generation approach replaced Pb-(OAc) 4 with an iodine(III) oxidant. In the event, treatment of (+)- 48 with PhI(OAc) 2 gave ortho-quinone like intermediate 49 . After workup and heating in toluene 49 underwent intramolecular Diels−Alder cycloaddition to give (+)- 50 . Two challenges remained: (1) adjustment of the A ring phenol and the C 6 carbon to their proper oxidation states and (2) closure of the piperidine D ring. Rearomatization followed by addition of TFA provided a phenolic amine that was tosylated to give bis-tosylate (−)- 51 . At this stage, the first and second-generation syntheses intercepted, thus completing the formal synthesis of ent-hydromorphone ( ent- 5)

Hudlicky’s Second Generation Chemoenzymatic Synthesis of ent-Hydromorphone
3.4.2. Hydrocodone.
Hydrocodone (7) is commonly found in combination with acetaminophen in the brand named analgesic Vicodin. Synthetically, it serves as an intermediate in the synthesis of codeine and morphine. 42 , 43
The survey begins with Rice’s racemic effort which is regarded as the approach most amenable to industrial scale up ( Scheme 4A ). 44 The synthesis began with condensation of carboxylic acid 52 and amine 53. The amide, thus obtained, was treated with POCl 3 to initiate a Friedel−Crafts like acylation event completed by the reduction of the intermediate imine. 45 Selective Birch reduction of the less electron rich C ring was followed by N-formylation to give formamide (±)-55. Methyl enol ether hydrolysis and concomitant ketalization set the stage for C 1 electrophilic aromatic bromination. Acidic hydrolysis of the acetal gave cyclization precursor (±)-56. Exposure of (±)-56 to NH 4 F·HF in TfOH provided cyclization product (±)-57. The C 1 bromide in the preceding step is critical to obtaining the desired cyclization product as previous studies indicate a high propensity for para cyclization at the expense of the desired ortho closure. 46 , 47 Next, hydrolysis of (±)-57 revealed the free secondary amine. α -bromination was followed by a 5-exo-tet ring closure to establish the E ring. Finally, reductive amination was accompanied by aryl bromide removal to give (±)-hydrocodone (7).

Total Synthesis of Hydrocodone from the Rice and Mulzer Labs
Mulzer and Trauner published a synthesis of (−)-hydrocodone ( Scheme 4b ). 48 , 49 Their effort began with aldol condensation of (±)-58 and methyl formate. After Robinson annulation with MVK, treatment with KOH cleaved the formate via a retro-Claisen reaction. Next, the racemic enone (±)- 59 was resolved using chiral chromatography to give (−)- 59 . 1,4-addition of a Grignard-copper(I) species was followed by trapping with TMSCl to give a transient silyl enol ether. This species was reacted with NBS to give α -bromo ketone (−)- 60 . Heating of (−)- 60 at 140 °C led to closure of the E ring. Following ketalization, a hydroboration, quenched with basic H 2 O 2 installed a distal primary alcohol. Subsequent dechlorination with Raney nickel gave intermediate (−)- 61 . After amine installation via Mitsunobu reaction with protected methylamine, a point of unsaturation was introduced via radical benzylic bromination and elimination. Dissolving metal reduction liberated the amine and set in motion reductive closure of the D ring. Ketal hydrolysis gave (−)-hydrocodone (7).
3.4.3. Oxycodone.
Oxycodone (8) is structurally identical to hydrocodone (7) sans C 14 oxygenation. Industrially, (8) is accessed through semisynthesis from (−)-thebaine (4) through a redox sequence ( Scheme 5 ). 50 , 51

Conversion of (−)-Thebaine to (−)-Oxycodone
The Fukuyama lab reported the first total synthesis of oxycodone that circumvents thebaine as an intermediate ( Scheme 6 ). 52 Starting from aryl bromide 65 , a Fagnou palladium catalyzed direct acylation closed the B-ring to give tricycle 66 . A further three step sequence provided phenol 67 . In a key-step, oxidative dearomatization in the presence of PhI(OAc) 2 proceeded through oxonium intermediate 68 to give C 14 hydroxylated product 69. Next, chemoselective hydrogenation with Wilkinson’s catalyst provided enone 70. At this stage, acylation of the C 14 alcohol with methyl malonyl chloride followed by exposure to Cs 2 CO 3 resulted in intramolecular Michael addition to form the benzylic quaternary carbon. Subsequent Krapcho decarboxylation and deprotection generated phenol 71 . After five additional steps amide 72 was treated with PhI(OAc) 2 to induce an oxidative Hofmann rearrangement. The amine, resulting from aqueous hydrolysis of the intermediate isocyanate, ring opened the neighboring lactone to reveal the C 14 alcohol and establish the piperidine D ring. A final two step sequence converted 73 to (−)-oxycodone (8).

Fukuyama’s 2014 Total Synthesis of (−)-Oxycodone
3.4.4. Buprenorphine and Naloxone.
Buprenorphine (10, Subutex) and naloxone (9, Narcan) are semisynthetic opiates used to treat drug dependence or overdose, respectively. Buprenorphine is a partial μ -receptor agonist often prescribed as an alternative to methadone to treat opioid dependence. It has less abuse potential than methadone, a full μ -receptor agonist, 53 and is also sold as a combination therapy with naloxone under the brand name Suboxone to further deter abuse. Naloxone is a μ -receptor antagonist that is United States Food and Drug Administration (FDA) approved for emergency treatement of known or suspected opioid and opiate overdose. 54 It is interesting to note that while naloxone is a receptor antagonist the majority of opiates are receptor agonists.
Synthetically, buprenorphine is made through semisynthesis from known opiate precursors. Starting from (−)-oripavine (3), amine alkylation and subsequent thiol mediated N-demethyla-tion gave diene (−)-74 ( Scheme 7 ). 55 An endo selective Diels− Alder cycloaddition with MVK followed by Grignard addition and hydrogenation provide (−)-buprenorphine (10). The observed diastereoselectivity of the Grignard addition can be rationalized by invoking model (75) in which the oxygens of the carbonyl and proximal methyl ether coordinate to magnesium, locking the carbonyl in place for top-side attack.

Synthesis of Buprenorphine
Similarly, the Hudlicky group recently disclosed a semisyn-thesis of (−)-naloxone (9) from (−)-oxymorphone (6) ( Scheme 8 ). 56 The synthesis commenced with acetate protection of the phenolic alcohol followed by amine oxidation. Next, dehydration with the Burgess reagent gave imine 77. While disfavored, 5-endo-trig ring closure at this stage would provide oxazolidine 79. One could argue that an alternative mechanism featuring a favored 5-exo-tet cyclization (78) is more likely. In either case, oxazolidine 79 is next protected as its dimethyl acetal to allow for clean oxazolidine opening with a vinyl Grignard reagent. Hydrolysis of the acetal protecting group reveals (−)-naloxone (9).

Hudlicky’s 2013 Synthesis of (−)-Naloxone
3.5. Illicit Semisynthetic Opiates.
While most opiate abuse is enabled through prescriptions, some are illicitly synthesized for recreational use.
3.5.1. Heroin.
First synthesized from morphine in 1874, the Bayer Company of Germany introduced heroin for medical use in 1898. Physicians remained unaware of its addictive potential for years, and, in 1903, heroin abuse had risen to alarming levels in the United States. Heroin was made federally illegal in 1924. Later, the Counter-Narcotics Police of Afghanistan (CNPA) offered German authorities the opportunity to observe and document the illicit synthesis of heroin ( Scheme 9 ). 57 The synthesis began with the isolation of morphine base from raw opium (82). Treatment of raw opium with lime (calcium oxide) and hot water resulted in a biphasic mixture in which the morphine base (1) resided in the lower aqueous layer. Siphoning of the aqueous layer into a fresh barrel followed by treatment with solid ammonium chloride resulted in the precipitation of morphine base. Next, the solid was treated with approximately 5 equiv of Ac 2 O to convert morphine freebase to heroin. Addition of Na 2 CO 3 neutralized the AcOH formed during the quench, precipitating brown heroin base in the process. Purification over activated carbon and precipitation with aq. NH 3 gave white heroin which was treated with HCl and acetone to give heroin hydrochloride (11·HCl).

Illicit Synthesis of Heroin from Raw Opium
3.5.2. Krokodil.
(−)-Desomorphine (12) or krokodil (from the Russian word meaning crocodile) became popular in Russia around 2003 and is a opioid synthesized from codeine and other readily obtained materials. 58 Over the past 15 years krokodil has slowly spread across Europe. It has also been sensationalized in a number of media reports as a drug that “turns the user into a zombie”. Indeed, krokodil can leave abusers disfigured via skin discoloration that results from cutaneous infection, gangrene, and necrosis. 1 The synthesis of krokodil begins with extraction of codeine containing tablets using alkaline pipe cleaner, gasoline, and HCl ( Scheme 10 ). 59 The resulting hydrochloride salt (2·HCl) is then reduced with I 2 and red phosphorus through a Nagai Type reaction.

Illicit Synthesis of Krokodil
4. THE OPIOIDS
4.1. historical perspective..
The term opioids was originally coined in the 1960s by George H. Acheson to “refer to any chemical with morphine-like properties.” These compounds were originally synthesized to develop both less addictive and more powerful analgesics. From this research, compounds like fentanyl (84) emerged. While all of the opioids are addictive, some are larger targets for abuse such as Demerol (83) and methadone (85), while others are less abused due to their high potency and increased likelihood for overdose.
4.2. Structure.
Based on the morphine rule, opioids have similar (or stronger) effects as the opiates due to their masked structural homology, vida supra. Thus, each molecule features a tertiary amine linked to an aryl ring ( Figure 2 ).

Structures of various opioids.
4.3. Demerol.
Demerol (83, also known as meperidine or pethidine) was first synthesized in 1939 and advertised as a “less addictive form of morphine” throughout the mid-1900s. However, a high level of dependence develops after initial use. Celebrities ranging from Elvis Presley to Michael Jackson have infamously suffered from Demerol addiction. In 1939, the Eisleb lab patented a synthesis of piperidine compounds, where di( β -halogenalkyl)-amine 87 is treated with benzyl cyanide. Cyclization is accompanied by nitrile hydrolysis to produce Demerol 83 ( Scheme 11 ). 60 Many of the current industrial syntheses use this method.

Eisleb’s 1939 Synthesis of Demerol
While lacking the practicality of Eisleb, Hite’s 1959 synthesis, featuring a quasi-Favorskii rearrangement, provides an interesting alternative ( Scheme 12 ). 61 Starting with monochloroketone 89, available in six steps from isonicotinic acid, treatment with NaOH induced the desired rearrangement, albeit in modest yield as the reaction’s minor product. Further inspection reveals that either α -phenyl or α -hydroxyl migration can occur (transition states 90 and 92, respectively). Although not directly applicable, comparison of the A-values for the phenyl (3.0 kcal mol − 1 ) and hydroxyl (≤1.0 kcal mol − 1 ) substituents prove instructive in evaluating the relative amount of diaxial-like strain felt by each group when positioned antiperiplanar to the exiting chloride. Returning to the synthesis, Fischer esterification of 91 gives Demerol (83).

Hite’s Synthesis of Demerol
4.4. Fentanyl.
In 1953, Paul Janssen set out to develop a new analgesic that would improve upon morphine and Demerol. 62 Hypothesizing that increased lipophilicity would increase CNS penetration and potency, Janssen and co-workers began modifying Demerol. 63 These efforts led to the synthesis of fentanyl (84) through a five step process involving reductive amination, acylation, and amine deprotection/alkylation ( Scheme 13 ). 64 Though first brought to the market as an intravenous analgesic, fentanyl soon became a common anesthetic for cardiac and vascular surgeries. Later developments allowed for administration via transdermal patches, buccal lozenges (lollipops), and intranasal sprays. 63 Fentanyl is a synthetic analgesic that is 50−100 times more potent than morphine. Given its powerful pain-relieving effects, and its ease of synthesis, fentanyl is often mixed with heroin and other drugs. In many parts of the United States and Canada, fentanyl has all but replaced heroin, unbeknownst to the user. 65

Janssen Company’s 1965 Synthesis of Fentanyl
4.5. Methadone.
In 1938, Bockmühl and Erhart synthesized compound VA 10820 that exhibited stronger analgesic effects than Demerol. Later named methadone (85), this compound is prescribed to treat opiate addiction. As methadone itself is highly addictive, many users cycle through the use of both opiates and opioids in an attempt to break dependence. The original synthesis of methadone (85) starts with racemic 1-dimethylamino-2-propanol (97) ( Scheme 14 ). Reaction with sodium diphenyl acetonitrile produces amino-nitrile (±)-99 via intermediate (±)-98. A Grignard addition concludes the synthesis to produce (±)-methadone (85).

Synthesis of (±)-Methadone
5. THE BENZODIAZEPINES
5.1. historical perspective..
Research to expand the ring of quinazoline-3-oxides in the late 1950s led to the development of the benzodiazepines. Leo Sternbach investigated the heterocycles of benzheptoxdiazines, known in the German literature in the late 1800s as acylindazoles, in order to replace the use of the highly addictive barbiturates. 66 During his research, Sternbach made a library of compounds that were not assayed until a colleague rediscovered them ca. 2 years later. The molecules showed remarkable activity as sedatives and sparked a major development campaign into this new class of compounds. 67 A series of substituted benzodiazepines were synthesized and marketed as the antianxiety medications readily prescribed today. Others, like flunitrazepam ( 104 ), affect cognitive function and promote anterograde amnesia, leading to abuse as the “date rape” drug commonly known as a “Roofie”. After some time, it would be demonstrated that the benzodiazepines were as addictive as the barbiturates they hoped to replace. Nonetheless, the benzodiazepines offered a new chemical scaffold that medicinal chemists continue to manipulate today.
5.2. Structure.
Benzodiazepines are named according to their fusion of the benzene and diazepine ring systems ( Figure 3 ). Benzodiazepine drugs are most commonly 1,4-substituted and can be differentiated based on modifications at the C 2′ , C 4′ , and C 7 positions.

Structures of various benzodiazepines.
5.3. Valium.
After the discovery of the first benzodiazepine ( 100 , Librium), diazepam ( 101 , Valium) was synthesized and demonstrated to be more potent. Valium would ultimately become the top selling drug in the United States from 1968 to 1982 but eventually received backlash as negative effects on cognition started to surface. 68 Beginning with ortho -amino-benzophenone ( 108 ), quinazoline-3-oxide ( 105 ) was generated in three steps. 69 , 70 After treatment with NaOH, 105 underwent ring expansion to give the cyclic 7-membered lactam 107 which could be elaborated to Valium ( 101 ) in two steps ( Scheme 15 ). Subsequent optimization reduced the synthesis from six steps to two ( Scheme 16 ). 66 , 68 This route began with condensation of ortho -aminobenzophenone ( 108 ) with glycine ethyl ester to establish the benzodiazepine ring system. Amide methylation completes the synthesis.

Sternbach’s Synthesis of Valium

Synthesis of Valium
The Gates lab achieved the first academic synthesis of Valium in 1980 ( Scheme 17 ). 71 Isatoic anhydride 109 was converted to 1,4-benzodiazepine-2,5-dione 110 via nucleophilic attack of the amine and subsequent decarboxylation and cyclization. Acetylation and Grignard addition resulted in aminobenzophe-none derivative 111 which yielded Valium ( 101 ) upon deacetylation and treatment with NaHSO 4 .

Gates’s Synthesis of Valium
5.4. Xanax.
The Upjohn Company patented the first synthesis of alprazolam ( 103 , Xanax) in 1971 ( Scheme 18a ). 72 , 73 Starting with quinoline derivative 112 , treatment with hydrazine gave the corresponding 2-hydrazinyl quinoline (not pictured) via an S N Ar (nucleophilic aromatic substitution) mechanism. Installation of the 1,2,4-triazole was realized after reaction with (EtO) 3 CCH 3 to give 113 . Treatment with O 3 followed by oxidative workup with aq. NaI and AcOH gave ketone 114 in good yield. Mechanistically ( Scheme 18b ), one can envision that collapse of secondary ozonide (±)- 115 could occur after iodide attack to yield aldehyde 116 and an equivalent of hydroiodite (IO − ). Reduction of this +1 iodine species could occur in situ under the influence of iodide and acid to give molecular iodine (I 2 ) which could in turn serve to oxidize 116 . After deprotonation of 117 , resonance stabilized acylium cation 118 is generated. Cation quenching with water would give carboxylic acid 120 which, following heterocycle protonation ((±)- 121 ), can undergo decarboxylation to yield final product 114 . Returning to the synthesis, hydroxymethylation of 114 with paraformaldehyde, formation of the chloride, and subsequent displacement with NH 3 provided an amine that cyclizes after condensation to give Xanax ( 103 ).

Upjohn Company’s 1971 Synthesis of Xanax
6. THE STIMULANTS
6.1. historical perspective..
Stimulants promote wakefulness and decrease fatigue. Compounds in this category range from caffeine to cocaine, amphetamine, ephedrine, and MDMA (ecstasy).
6.2. Structure.
Cocaine ( 122 ) is a bicyclic tropinone alkaloid, which represents one of the more complex stimulant structures ( Figure 4 ). Amphetamine, ephedrine, methamphetamine, and methylphenidate are classified as phenethylamines. MDMA is a phenethylamine derivative containing a 3,4-methylenedioxy bridge.

Structures of various stimulants. a Bronkaid is combination of (−)-ephedrine and guaifenesin. b The active ingredient in Ritalin and Concerta is (±)-methylphenidate.
6.3. Cocaine.
The euphoric effects that result from chewing the leaves of the Erythroxylum coca plant have been known to humankind for millenia. 74 However, it was not until the mid-19th century that the coca-alkaloid cocaine ( 122 ) was isolated from this natural source. 75 , 76 Soon after, cocaine gained popularity in the United States as a local anesthetic, a cure for opiate use disorder, and an additive to cigarettes and soda. At the turn of the century, cocaine’s addictive nature emerged, and the United States responded with the 1914 Harrison Act to restrict the drug’s sale and distribution. This largely ended the country’s first cocaine epidemic.
From a synthetic perspective, cocaine’s compact bicyclic structure and biological activity has long attracted attention. The Willstatte r ̈ lab completed the first synthesis of (−)-cocaine ( 122 ) in 1903 ( Scheme 20 ). 77 , 78 This was a landmark moment in organic synthesis, as it involved the formation of the tropinone core and set a new precedent for the complexity that chemical synthesis could reach. The synthesis began with the transformation of cycloheptanone into cycloheptatriene ( 130 ). Treatment of 130 with Br 2 and (CH 3 ) 2 NH followed by chemoselective reduction provided unsaturated amine 131 . After dibromination of the remaining olefin, intramolecular bromide displacement provided bicyclic bromide salt (±)- 133 , presumably through a transition state similar to 132 . Bromide elimination and amine monodemethylation gave olefin(±)- 134 which was subjected to bromination, hydrolysis, and oxidation to complete the synthesis of tropinone ( 135 ). Treatment of tropinone with Na in EtOH in the presence of CO 2 gave the α -carboxylic acid which was converted to methyl ester (±)- 136 under acidic conditions. Reduction with Na(Hg) and subsequent benzoylation afforded (±)-cocaine ( 122 ). A chiral resolution with D-tartaric acid allowed for the isolation of naturally occurring (−)-cocaine ( 122 ).

Willstätter’s Synthesis of (−)-Cocaine from 1903
While trying to elucidate the biosynthetic pathways responsible for the synthesis of the tropinone scaffold, Sir Robert Robinson demonstrated a simplified synthesis of tropinone ( 135 ) in 1917 ( Scheme 19 ). 79 , 80 In a one-pot procedure, aqueous methylamine was added to a solution of succinaldehyde ( 137 ) and calcium acetonedicarboxylate ( 138 ). Following condensation of methylamine with aldehyde 137 , consecutive Mannich reactions gave tropinone ( 135 ). This reaction laid the groundwork for Robinson’s proposal for the biosynthetic pathway of cocaine and similar alkaloids, an area of research that eventually earned him the Nobel Prize in Chemistry in 1947.

Robinson’s 1917 Synthesis of Tropinone
Unsurprisingly, cocaine’s intricate bicyclic structure has continued to attract the interest of synthetic chemists long after the seminal efforts of Willstätter and Robinson. Rappaport and co-workers reported an enantioselective synthesis of (−)-cocaine ( 122 ) in 1998 ( Scheme 21 ). 81 Using the chiral pool as its source of asymmetry, the synthesis began with the Eschenmoser sulfide contraction of racemic dibenzyl malate ( 139 ) and D-glutamic acid derived chiral thiolactam (−)- 140 . Treatment of (±)- 139 with Tf 2 O afforded the corresponding α -triflate which was displaced after addition of the thiolactam. Addition of PPh 3 as a sulfur scavenger followed by the introduction of a tertiary amine base completed the one pot reaction sequence to give vinyl amine (+)- 141 as an inconsequential mixture of E and Z isomers. Catalytic hydrogenation of (+)- 141 resulted in debenzylation and was accompanied by decarboxylation of the resulting vinylic acid carboxylic acid affording a chiral amino acid which, upon reprotection of the liberated carboxylic acid and amine functionalities, was transformed into (+)- 142 . KHMDS mediated Dieckmann cyclization gave bicyclic β -keto ester (+)- 143 . Nucleophilic decarboxylation followed by BamfordStevens reduction gave tropene intermediate (+)- 144 which underwent an exo selective [3 + 2] dipolar cycloaddition to give 4,5-dihydroisoxazole (−)- 145 . Ester hydrolysis followed by thermal decarboxylation and concomitant cleavage of the N−O bond gave β -hydroxyl cyanide (+)- 146 which was elaborated in six further steps to (−)-cocaine ( 122 ).

Rapoport’s 1998 Synthesis of (−)-Cocaine
In regard to its illicit use, cocaine reemerged as a highly abused drug in the United States in the latter half of the 20th century. It is primarily consumed in one of two forms: powder cocaine and crack cocaine ( Scheme 22 ). Powder cocaine, better chemically defined as the HCl salt of (−)-cocaine ( 122 ·HCl), is used via injection or insufflation. While this is the form in which cocaine is most commonly isolated, the free amine commonly known as crack cocaine is far more addictive. Crack cocaine reaches the brain quicker than the hydrochloride salt and produces a more intense high. The name “crack” is derived from the sound this form of cocaine makes when burned. This form of cocaine is illicitly made from the cocaine salt ( Scheme 22 ). In this process, powder cocaine ( 122 ·HCl) is treated with aq. NaHCO 3 . After heating, the free base ( 122 ) separates from the water into an oil that can be solidified.

Conversion of Powder Cocaine to Crack Cocaine
6.4. Phenethylamines.
The phenethylamines represent one of the most widely abused and illicitly produced classes of drugs today. The structural backbone of the class is seen in amphetamine ( 123 ), which features a single stereocenter and a key primary amine ( Figure 4 ). Their structural similarity makes them easy to interconvert and alter. In this section, we describe general synthetic techniques used to produce these compounds and detail their illicit production.
6.4.1. Amphetamine.
Amphetamine ( 123 ) is a stimulant that increases wakefulness and focus while decreasing fatigue and appetite. Physicians often prescribe amphetamine as a 1:1:1:1 mixture, by weight, of four salts ( Figure 5 ) to treat attention deficit hyperactivity disorder (ADHD). This mixture, sold under the brand name Adderall, and other stimulants prescribed to treat ADHD, such as (+)-lisdexamfetamine (Vyvanse, 124 ) and (+)-methylphenidate (Focalin, Ritalin, Concerta, 128 ), are often abused by college students as “study drugs”.

Structures of the four amphetamine salts that constitute Adderall.
Synthetically, Edeleanu first prepared amphetamine ( 123 ) in 1887 and subsequently adapted the synthesis to access both ephedrine ( 125 ) and methamphetamine (127). 82 However, interest in developing these compounds for use as drugs did not emerge until the early 1960s, at which point several syntheses made racemic amphetamine widely available. For example, in 1968, Belovsky and co-workers demonstrated a simple synthesis of (±)-amphetamine ( 123 ) from phenylacetone ( 151 ) and formamide ( Scheme 23 ). 83 The synthesis began with the condensation of phenylacetone ( 151 ) and formamide under Leuckhart conditions to give (±)- 152 . Subsequent hydrolysis yielded (±)-amphetamine ( 123 ), completing the synthesis in two simple transformations. A variety of other traditional syntheses of racemic amphetamine exist and use key transformations such as the Ritter reaction, Friedel−Crafts acylation, Knoevenagal condensation, Curtius rearrangement and Wolff rearrangement. 84

Belovsky’s Synthesis of (±)-Amphetamine
While effective as a racemate, studies with enantiopure amphetamine have revealed that (+)-amphetamine demonstrates higher potency in the CNS while (−)-amphetamine has a larger effect on cardiovascular activity. As such, many have put forth efforts to access amphetamine in enantiopure form either through chiral resolution or enantioselective synthesis. These synthetic efforts engender chirality through the use of chiral starting materials, asymmetric hydrogenations, or enzymatic transformations. 84 Perhaps most prominent of these methods is Yamaguchi’s use of chiral aziridines ( Scheme 24 ). 85 Beginning with chiral building block L-alinol ( 153 ), phosphonate protection followed by mesylation provided 154 . Next, this intermediate underwent base promoted intramolecular displacement to give chiral aziridine ( 155 ). Treatment of the aziridine with a mixture of PhMgBr and catalytic CuCl resulted in nucleophilic attack at the less substituted carbon. Finally, acid mediated removal of the N -phosphonate revealed enantiopure (+)-amphetamine ( 123 ). A number of variations on this method have been reported. 86

Yamaguchi’s Synthesis of (+)-Amphetamine
6.4.2. (Pseudo)ephedrine.
Indiscriminate hydroxylation of the benzylic position of amphetamine yields two enantiomeric sets of diastereomers ( Figure 6 ). The erythro diastereomers constitute the ephedrines ( 125 ) while the threo diastereomers comprise the pseudoephedrines ( 126 ). Medicinally, the erythro and threo diastereomers demonstrate different activities. Ephedrine stimulates the central nervous system, promoting wakefulness and decreased appetite, while pseudoephedrine acts on the peripheral nervous system and is used as a nasal decongestant sold under the brand name Sudafed.

Comparison of ephedrine and pseudoephedrine stereo-chemistry. The medicinally used enantiomer of each compound is depicted.
6.4.3. Methamphetamine.
Mono- N -methylation of amphetamine ( 123 ) yields methamphetamine ( 127 ), a commonly abused drug with street names such as “meth” and “crank”. Due to the relative ease with which it can be synthesized, many clandestine laboratories manufacture this highly addictive drug. However, such synthetic work is not without danger as methamphetamine production frequently leads to “lab” explosions and house fires. The impact of such accidents as well as the socioeconomic ramification of methamphetamine addiction make plain the motivation behind the controlled nature of pseudoephedrine ( 126 ) containing medications, methylating reagents, and reducing agents.
Synthetically, methylation of amphetamine ( 123 ) with dimethyl sulfate can generate methamphetamine ( 127 ); however, the high propensity for these conditions to yield dimethylamphetamine makes this method less appealing. 87 The mono-N-methylated ephedrine ( 125 ) and pseudoephedrine ( 126 ) (jointly referred to as (pseudo)ephedrine) are more common starting materials as various routes exist for the reduction of (pseuo)ephedrine to methamphetamine (127). The most common strategy employes HI (Nagai route) or Birch reduction.
In recent years, a modification of the Birch reduction referred to as “shake and bake” has become the method of choice for illicit methamphetamine synthesis ( Scheme 25 ). This method usually starts with (−)-pseudoephedrine containing decongestants. The pills are crushed and subsequently added to a plastic bottle. NH 4 NO 3 , Li 0 , solid NaOH, and xylenes, chemicals available to the layperson from the household goods highlighted in red, are added. Next, iterative cycles of shaking and venting are used. After gas evolution ceases, the reaction is filtered and acidified with gaseous HCl to give methamphetamine (127) as its hydrochloride salt.

Illicit Synthesis of Methamphetamine via the “Shake and Bake” Method
Commonly referred to as “ecstasy” or “Molly”, MDMA ( 129 ) is a strong stimulant that prevents synaptic reuptake of serotonin, dopamine, and norepinephrine. It is so effective that, when used repeatedly, it can deplete the number of neurotransmitters available in the brain, producing a mental and physical crash that results in long-term depression. MDMA also affects the temperature-regulating mechanisms of the brain with users commonly dying from hyperthermia and hyponatremia (depletion of sodium content). Merck patented the first commercial MDMA ( 129 ) synthesis in 1912 ( Scheme 26 ). 88 Beginning with safrole ( 156 ), Markovnikov hydrobromination of the terminal olefin gives the corresponding secondary bromide. Finkelstein reaction provides a secondary iodide that is displaced with methylamine to give (±)-MDMA ( 129 ).

Merck’s 1912 Synthesis of (±)-MDMA
The Shulze lab recently published an MDMA synthesis that features a Curtius rearrangement ( Scheme 27 ). 89 Treatment of helional ( 157 ) with sodium propionate and propionic anhydride resulted in a Perkin reaction to give unsaturated carboxylic acid 158 . Olefin hydrogenation followed by acyl chloride synthesis gave (±)- 159 . At this point, generation of the acyl azide and subsequent Curtius rearrangement provided an isocyanate. Exposure of this intermediate to t -BuOK gave the corresponding Boc carbamate. N-Methylation followed by acid mediated Boc deprotection provided (±)-MDMA ( 129 ) which was isolated as its oxalate salt.

Schulze’s 2010 Synthesis of (±)-MDMA
In the hands of the clandestine chemist, illicit MDMA synthesis often begins with distillation of safrole ( 156 ) from the more readily available sassafras oil ( Scheme 28 ). Subsequent Wacker−Tsuji oxidation using PdCl 2 (available as a photograph developing agent) yields the ketone MDP2P ( 161 ). Subsequent reductive amination with methylamine utilizing any one of a variety of reducing agents (e.g., aluminum amalgam, available from the treatment of aluminum foil with a mercury(I) salt) provides (±)-MDMA ( 129 ). An interesting variation on this reaction bypasses the need for the highly regulated methylamine, instead using the more readily available nitromethane to generate methylamine in situ upon addition to the reaction’s reducing environment.

Illicit Synthesis of MDMA
7. THE HALLUCINOGENS
7.1. historical perspective..
Naturally occurring hallucinogens have been used for thousands of years as recreational drugs. Consumed for their spiritual and cognitive effects, many of these compounds structurally mimic neurotransmitters such as dopamine ( 162 ) and serotonin ( 163 ) ( Figure 7 ) and have significant impacts on the CNS with users often claiming to have “spiritual awakenings” under their influence.

Structures of dopamine and serotonin, two neuro-transmitters.
In response, a number of countries, have challenged their legality over the last century. In particular, during the mid-1960s counterculture movements in the United States, many Americans campaigned for the decriminalization of (+)-lysergic acid diethylamide ( 164 , LSD).
7.2. Structure.
As noted above, many hallucinogenic compounds share structural homology with either dopamine ( 162 ) or serotonin ( 163 )(Figures )(Figures7 7 and and8). 8 ). This is most readily seen in the structures of mescaline ( 167 ) and N,N -dimethyltryptamine ( 168 , herein referred to as DMT), respectively. However, careful examination of the structure of LSD ( 164 ) also reveals a resemblance to serotonin. On the other hand, ketamine ( 165 ) and phencyclidine ( 166 , herein referred to as PCP) do not resemble either of the aforementioned neurotransmitters yet still elicit hallucinogenic effects.

Structures of various hallucinogens. a Resemblance to serotonin is highlighted in red.
In the midst of a synthetic effort directed at accessing various amide analogues of lysergic acid, Albert Hofmann unwittingly constructed a derivative that would quickly permeate the world as a hallucinogen: LSD ( 164 ). However, it was not until 5 years later that Hofmann would accidentally ingest a small amount of LSD and discover the psychedelic properties of this semisynthetic ergot alkaloid. 90 While initially used by the medical community, LSD soon became a prominent illegal recreational drug. 91 Structurally, LSD shares a tetracyclic skeleton with clinically relevant ergot alkaloids such as ergometrine ( 170 ) and ergotamine ( 171 ) ( Figure 9 ). 92

Structures of various ergot alkaloids.
Industrial access to clinically relevant ergot alkaloids relies on direct isolation from fermentation or field cultivation. 93 , 94 Similarly, one can imagine LSD resulting from the amination of lysergic acid ( 169 ) or arising from the hydrolysis of drugs such as ergometrine ( 170 ) or ergotamine ( 171 ). Consequently, the syntheses covered in the following section revolve around the synthesis of lysergic acid as a formal synthesis of LSD.
Woodward and co-workers published the first total synthesis of (±)-lysergic acid ( 169 ) in 1954 ( Scheme 29 ). 95 , 96 Beginning from known dihydroindole derivative (±)- 172 , acyl chlorination followed by AlCl 3 mediated Friedal−Crafts acylation closed the C ring to furnish (±)- 173 , a 2,2- α -dihydro derivative of Uhle’s ketone. Ketone (±)- 173 was treated with pyridinium tribromide to afford the corresponding α -bromide which could be displaced with methylamino ketal 174 . Subsequent acid hydrolysis of the ketal and benzamide protecting groups revealed diketone (±)- 175 which, upon treatment with NaOMe, underwent aldol condensation to close the D ring and give (±)- 176 . Six subsequent transformations completed the first total synthesis of (±)-lysergic acid 169 .

Woodward’s Approach to (±)-Lysergic Acid
Fukuyama’s 2013 asymmetric synthesis of (+)-lysergic acid was equally instructive ( Scheme 30 ). 97 Chiral β -hydroxyl hydrazide (−)- 177 , the product of Evans aldol condensation and subsequent auxiliary cleavage with hydrazine, was treated with t -BuONO to generate acyl azide 178 . In situ Curtius rearrangement afforded isocyanate 179 which underwent intramolecular trapping to produce oxazolidinone (−)- 180 . After three transformations, ring closing metathesis installed the A ring, giving (−)- 182 . Next, a Heck cyclization closed the B ring to give (−)- 184 after β -hydride elimination. Functional group interconversions over the course of 11 steps transformed this compound into the final product, (+)-lysergic acid (169). It is worth noting the authors’ judicious control of the β -hydrogen stereochemistry in intermediate 183; the syn positioning of this hydrogen and the palladium species enabled β -hydride elimination, demonstrating a firm command of mechanistic understanding.

Fukuyama’s Synthesis of (±)-Lysergic Acid
In a similar vein, Fujii and Ohno published a 2011 formal synthesis of (+)-lysergic acid ( 169 ) featuring an amino-palladation cascade that established the C and D rings in a single step ( Scheme 31 ). 98 Starting with enyne 186 , available in four steps from 4-bromoindole ( 185 ), Shi epoxidation utilizing catalyst 187 afforded epoxide 188 which was taken forward as a mixture of diastereomers. Silyl protection of the primary alcohol followed by Zn(OTf) 2 mediated reductive oxirane opening yielded ynol 189 . At this stage, the authors established the allene needed for the amino-palladation cascade via a Movassaghi modified Myers allenation; 99 stereoselective formation of 192 occurred, presumably, through the concerted Alder-Ene reaction of intermediate 191 . With the allene in hand, TBAF deprotection of the primary alcohol and subsequent treatment with Pd(Ph 3 ) 4 and K 2 CO 3 , cyclization conditions previously established by the authors, 100 yielded tetracycle 194 . The amino-palladation precursor and the resulting product ( 194 ) intersected with a previously disclosed synthesis of (+)-lysergic acid by the authors. 100

Fujii and Ohno’s Synthesis of (±)-Lysergic Acid
7.4. Ketamine.
A molecule of many uses, ketamine ( 165 ) has long served as an anesthetic, analgesic, and recreational drug known on the street by the name “Special K.” 101 ParkeDavis and Company disclosed the seminal synthesis of (±)-ketamine in a 1956 patent ( Scheme 32 ). 102 The synthesis began with o -chlorobenzonitrile ( 195 ) which yielded ketone 196 after treatment with a cyclopentyl Grignard reagent. Subsequent α -bromination followed by imine formation and bromide hydrolysis gave α-hydroxyl imine 197 , the precursor for the synthesis’ key pinacol-like rearrangement. In the event, heating 197 in decalin resulted in [1,2] migration to give (±)-ketamine ( 165 ).

Parke-Davis and Company’s 1956 Synthesis of (±)-Ketamine
More recently, Zhang and co-workers reported a racemic ketamine synthesis that started from ketone (±)- 198 ( Scheme 33 ). 103 Direct α -nitration mediated by ceric ammonium nitrate (CAN) and Cu(OAc) 2 provided α -nitro ketone (±)- 199 . Nitro reduction and subsequent reductive amination with aqueous formaldehyde yielded (±)-ketamine ( 165 ).

Zhang’s Synthesis of (±)-Ketamine
While both of these syntheses provide facile access to (±)-ketamine, research has demonstrated that (−)-ketamine exhibits greater potency than the corresponding (+)-ketamine antipode in regards to anesthesia and analgesia. 104 As such, methods for accessing (−)-ketamine are gaining importance. Kiyooka recently published the first 105 and second 106 asymmetric syntheses of (−)-ketamine ( 165 ).
Kiyooka’s second synthesis begins with an enantioselective Mukaiyama aldol condensation mediated by chiral oxazoborolidinone 201 with α,β -unsaturated aldehyde ( P/M )- 200 ( Scheme 34 ). The resulting enantioenriched (86% ee) β -hydroxyl ester ( P/M )- 202 was subjected to LiAlH 4 reduction and selective benzylation of the resulting primary alcohol. Acylation of the secondary alcohol with trichloroacetyl isocyanate and subsequent hydrolysis gave carbamate ( P/M )- 203 . After dehydration, the carbamate was converted to isocyanate (−)- 205 . Mechanistically, this reaction occurs via dehydration of the carbamate to form intermediate allyl cyanate ( P/M )- 204 which undergoes a stereoselective [3,3]-sigma-tropic rearrangement giving the isocyanate product with 1,3-chirality transfer. Finally, reduction of the isocyanate ( 205 ) to give the corresponding methylamine was followed by reductive ozonolysis to give enantioenriched (−)-ketamine ( 165 ) in 87% ee.

Kiyooka’s Second Generation Synthesis of (−)-Ketamine
While Kötz and Merkel reported the first synthesis of PCP ( 166 ) in 1926, 107 the compound was not fully investigated for is biological properties until the late 1950s when it was made available as an anesthetic (Sernyl). 108 Research demonstrated that the compound elicited the desired anesthetic effects but was also a strong hallucinogen. This promptly led to its discontinued use in 1965. The hallucinogenic effects, however, fueled abuse of PCP.
Traditional syntheses of PCP are based on common cyclohexanone precursors, making them optimal for conversion into clandestine syntheses. 109 , 110 One commonly used procedure is exemplified by Parke-Davis and Company’s 1965 synthesis of PCP ( Scheme 35 ). Beginning from cyclohexanone ( 206 ), reaction with aq. NaHSO 4 provided an addition product that was treated with aqueous piperidine and KCN to give PCC ( 207 ). PhMgBr addition to the imine intermediate, presumably formed in situ from 207 , provides PCP ( 166 ).

Parke-Davis and Company’s 1965 Synthesis of PCP
An alternative synthesis from Pons began with conversion of tertiary alcohol 208 to the corresponding tertiary azide ( Scheme 36 ). 111 Presumably, this substitution proceeds through an S N 1 manifold via a benzylic carbocation. Reduction of the azide provided a tertiary amine, which was elaborated to PCP ( 166 ) in good yield after dialkylation with 1,5-dibromopentane.

Pons’s Synthesis of PCP
Clandestine syntheses of PCP often proceed according to routes similar to that depicted in Scheme 34 and can be performed on large scales. One such manifestation is termed the “The Bucket Method” ( Scheme 37 ). 109 Following instructions seized from a clandestine PCP laboratory by the United States Drug Enforcement Administration (DEA), the starting materials are first divided into two buckets, Bucket A and Bucket B ; to Bucket A are added cyclohexanone ( 206 ) and Na 2 S 2 O 5 ( 209 , which can generate NaHSO 4 in situ when exposed to water), while piperidine ( 210 ) and an alkali cyanide salt are added to Bucket B . Addition of the contents of Bucket A to those of Bucket B (or vice versa) yields masked iminium species PCC ( 207 ). In a third bucket, Bucket C, bromobenzene ( 211 ) is added to magnesium turnings to generate PhMgBr. This Grignard reagent is then added to a solution of PCC in naphtha to give PCP ( 166 ). Because such processes can readily produce large batches of PCP, the DEA stringently monitors several related compounds, such as piperidine and potassium cyanide.

Illicit Synthesis of PCP by the “Bucket” Method
7.6. Mescaline.
Native Americans in Central and North America have used mescaline ( 167 ), the active component in the peyote cactus, for thousands of years in spiritual ceremonies. The “buttons” of the cactus are often dried and eaten or soaked in water to drink. Mescaline ( 167 ) is believed to mimic the neurotransmitter dopamine ( 162 ) (vide supra) and causes intense hallucinations and altered states of consciousness. Mescaline was first synthesized by Späth and co-workers in 1919 ( Scheme 38 ). 112 Beginning with eudesmic acid ( 212 ), treatment with SOCl 2 gave the corresponding acyl chloride. Reduction under Rosenmund’s conditions gave aldehyde 213 which could be elaborated to nitrostyrene 214 through a Henry reaction with nitromethane. With the molecule’s carbon skeleton established, successive reductions with Zn in AcOH and then with Na(Hg) provided mescaline ( 167 ). In 1950, Morin and co-workers reported a similar synthesis using LiAlH 4 to convert the nitrostyrene intermediate 214 directly into the primary amine in higher yields. 113

Spatḧ’s 1919 Synthesis of Mescaline
Tsao published a complementary mescaline synthesis in 1951 ( Scheme 39 ). 114 Beginning with eudesmic acid ( 212 ), a three step sequence of esterification, reduction, and chlorination yielded benzyl chloride 215 . Addition of the remaining aminomethylene unit was accomplished through Kolbe nitrile synthesis and LiAlH 4 reduction.

Tsao’s Synthesis of Mescaline
An entheogen is any psychoactive substance that induces a spiritual experience. Ayahuasca is a spiritual medicinal brew traditionally used in ceremonies by peoples of the Amazon river basin. DMT ( 168 ) is the active component in ayahuasca. 115 Its biosynthesis occurs within plants via the shikimate pathway. 116 This pathway is also responsible for the biosynthesis of the amino acid tryptophan as well as various indole alkaloid precursors. The direct precursor of DMT is tryptamine ( 216 ), which was first chemically synthesized by Shapiro and co-workers in 1956 ( Scheme 40 ). 117 The synthesis begins with an intermolecular Michael addition between diethyl malonate ( 217 ) and acrylonitrile ( 218 ) to give a product that cyclizes after Raney nickel reduction to yield (±)- 219 . A Japp− Klingemann reaction with benzenediazonium chloride gives hydrazine 220 which undergoes cyclization when exposed to polyphosphoric acid to give (±)- 221 . Amide hydrolysis and subsequent decarboxylation yielded tryptamine ( 216 ).

Shapiro’s Synthesis of Tryptamine
Conversion of tryptamine ( 216 ) to DMT ( 168 ) occurs under standard methylating conditions or through reductive amination ( Scheme 41 ). 118

Conversion of Tryptamine to DMT
8. CONCLUSION
A number of key lessons were learned during the assembly of this document. First, the field of psychoactive drug synthesis is rich with foundational strategies and tactics to prepare both polycyclic and linear alkaloids. While the seminal Gates synthesis was completed 65 years ago, the field remains vibrant with countless players distributed worldwide. Academic syntheses, however, are primarily instructional. Even in an era where synthetic approaches are arguably the shortest they have ever been, the use of total synthesis in matching the efficiency of direct isolation or semisynthesis is futile (at least in the case of the complex opium or coca alkaloids). Perhaps in time an “ideal” four to six-step synthesis will emerge that challenges plant isolation.
Second, it is clear that the early exercises of our synthetic education are showcased throughout psychoactive drug production. Whether it is the ability to produce a 5- or 6-membered ring (the latter via Diels−Alder technology) or introduce tertiary amines through reductive amination, basic organic chemistry maneuvers are critical to the production of psychoactive drugs, be it in the lab or in the garage.
A final point is rather distal in nature and focuses on the future of psychoactive drug production. Currently, the United States is overwhelmed by opioid use disorder. Since 2000, benzodiazepine, painkiller, and heroin deaths have all risen to record levels. While there are a number of social strategies in place to combat the problem, perhaps the solution lies at the hands of synthetic chemists and our ability to develop compounds that address pain and suffering while minimizing addiction.
ACKNOWLEDGMENTS
We’d like to thank Dr. Katherine Chong for critical feedback and the reviewers for their suggestions.
S.D.T. would like to acknowledge Vanderbilt University and the Institute of Chemical Biology for financial support. S.A.C. acknowledges support from the Vanderbilt Chemical Biology Interface (CBI) training program (T32 GM065086). E.D.H. acknowledges support from the Vanderbilt Chemical Biology of Infectious Diseases (CBID) training program (T32 AI112541).
ABBREVIATIONS
The authors declare no competing financial interest.
IMAGES
VIDEO
COMMENTS
Organic synthesis is a rate-limiting factor in drug discovery. The timelines required in industry determine whether complex molecules are practically accessible; continued investment in innovative ...
Learn how to design and synthesize new molecules for drug discovery and development. This chapter covers concepts, strategies, tactics, examples and tools of organic synthesis.
Drug synthesis is an expensive and prolonged process challenged by ethical disputes associated with in vivo models and limited outcomes. Current efforts have proved efficient for the production and modification of novel drug-delivery systems in the pharmaceutical industry. Contemporary drug-delivery systems have shown effective nanoparticle ...
Medicinal chemistry encompasses the design and synthesis of novel bioactive compounds that have the potential to become drugs. It typically involves cycles of iterative optimization of compounds ...
Drug discovery is the process through which potential new therapeutic entities are identified, using a combination of computational, experimental, translational, ... drug design and synthesis approaches, in vitro and in vivo pharmacological and toxicological evaluations, etc. These articles not only provided important information, but also ...
The concise synthesis of the antiplatelet drug tirofiban is an excellent example of how the pharmaceutical industry can readily use this methodology to facilitate drug discovery and development. As research continues to surge in this field, additional breakthroughs are anticipated, and these will likely change how molecules are designed and built.
Leads for drug discovery are frequently identified by screening collections of compounds available from synthesis or by isolation from natural sources. In that these collections or libraries of compounds may be large (> 100,000), high-throughput screening methods and equipment have been developed to facilitate the work.
Aromatic nitro compounds play a unique role in the synthesis of drugs and pharmaceutically oriented molecules. This field of organic chemistry continues to be in demand and relevant. A significant number of papers are published annually on new general methods for the synthesis of nitrodrugs and related biomolecules. This review is an analysis ...
This article highlights how the design and synthesis of such libraries have been enabled by modern synthetic chemistry and illustrates the impact of the resultant chemical probes and drug leads in ...
The Art of Drug Synthesis, were published in 2004 and 2007, respectively. They have been warmly received by the chemistry community. Here is our third installment in the Drug Synthesis series. This book has five sections. Section I, "Infectious Diseases" covers three drugs; Section II, "Cancer" reviews five drugs, three of which are kinase ...
An Introduction to Drug Synthesis explores the central role played by organic synthesis in the process of drug design and development. Written by an experienced and talented author to complement his existing An Introduction to Medicinal Chemistry, the book illustrates how organic synthesis makes important contributions throughout the drug design and discovery process - from the generation of ...
A review. New drugs are introduced to the market every year and each represents a privileged structure for its biol. target. These new chem. entities (NCEs) provide insights into mol. recognition and also serve as leads for designing future new drugs. This review covers the synthesis of 15 NCEs that were launched anywhere in the world in 2010.
Drug synthesis. Get an email alert for Drug synthesis Get the RSS feed for Drug synthesis; Showing 1 - 13 of 14 View by: Cover Page List Articles. Sort by: Recent Popular. Indenyl-thiazole and indenyl-formazan derivatives: Synthesis, anticancer screening studies, molecular-docking, and pharmacokinetic/ molin-spiration properties ...
Historically, the United States has been at the forefront of innovative drug research and development worldwide. This article aims to provide guidance for researchers involved in new drug research and development by presenting the clinical applications and synthesis methods of 29 newly approved drugs by the FDA in 2023.
A review. Parallel library synthesis is an important tool for drug discovery because it enables the synthesis of closely related analogs in parallel via robust and general synthetic transformations. In this perspective, we analyzed the synthetic methodologies used in >5000 parallel libraries representing 15 prevalent synthetic transformations.
Drug Synthesis. Another API for NSAIDs synthesis is the enantiopure S-isomer of mandelic acid (hereafter (S)-MA), which is considered an important intermediate for the synthesis of selective COX-2 inhibitors such as celecoxib (Celebrex®) and deracoxib (Deramaxx®). From: Biotechnology of Microbial Enzymes, 2017.
Following Contemporary Drug Synthesis and The Art of Drug Synthesis (Wiley, 2004 and 2007), two well-received works, is this new book that demystifies the process of modern drug discovery for practitioners and students. An enhanced introduction covers areas such as background, pharmacology, SAR, PK/PD, efficacy, and safety. Focusing on the advantages of process synthesis versus the discovery ...
Biocatalysis is constantly providing novel options for the synthesis of active pharmaceutical ingredients (APIs). In addition to drug development and manufacturing, biocatalysis also plays a role in drug discovery and can support many active ingredient syntheses at an early stage to build up entire scaffolds in a targeted and preparative manner.
The Art of Drug Synthesis illustrates how chemistry, biology, pharmacokinetics, and a host of other disciplines come together to produce successful medicines. The authors have compiled a collection of 21 representative categories of drugs, from which they have selected as examples many of the best-selling drugs on the market today. ...
Biocatalysis is constantly providing novel options for the synthesis of active pharmaceutical ingredients (APIs). In addition to drug development and manufacturing, biocatalysis also plays a role in drug discovery and can support many active ingredient syntheses at an early stage to build up entire scaffolds in a targeted and preparative manner. Recent progress in recruiting new enzymes by ...
Nature Reviews Drug Discovery - Advances in areas such as microfluidics-assisted chemical synthesis and biological testing, as well as in artificial intelligence systems, are increasingly providing...
Click Chemistry Mechanism. Click chemistry is a newer approach to the synthesis of drug-like molecules that can accelerate the drug discovery process by utilizing a few practical and reliable reactions. Sharpless and coworkers defined what makes a click reaction as one that is wide in scope and easy to perform, uses only readily available ...
In drug design, AI is used to predict the 3D structure of proteins, drug-protein interactions and drug activity, constructs molecules de novo. In pharmacology, AI is used to design specific molecules as well as multitarget drugs. In chemical synthesis, AI is able to design synthetic route, to predict reaction yield, to clarify reaction ...
Feb. 11, 2020 — After helping develop a new approach for organic synthesis -- carbon-hydrogen functionalization -- scientists are now showing how this approach may apply to drug discovery ...
Abstract. 53 New drugs including 38 chemical entities and 15 biologics were approved by the U.S. Food and Drug Administration during 2020. Among the marketed drugs, 34 new small molecule drugs and 4 new diagnostic agents with privileged structures and novel clinical applications represent as promising leads for the development of new drugs with ...
Protein synthesis is indispensable in cell proliferation; hence, we evaluated the protein synthesis activity using OPP, which was labeled on the C-terminus of nascent polypeptide chains . Strobilurin X decreased the cell fluorescent intensity labeled by protein synthesis for 2 h in A549 cells, indicating that strobilurin X inhibits protein ...
Despite their critical importance in drug development and biochemistry, efficiently synthesizing α-glycosyl azides has continued to pose significant challenges. In this report, we introduce a universal and practical radical reaction for the stereoselective synthesis of α-glycosyl azides using bench-stable allyl glycosyl sulfones as the donor.
Drug Design Discov. 16, 249-255 (2019). Article CAS Google Scholar Durgun, M. et al. Synthesis, characterisation, biological evaluation and in silico studies of sulphonamide Schiff bases.
First, the field of psychoactive drug synthesis is rich with foundational strategies and tactics to prepare both polycyclic and linear alkaloids. While the seminal Gates synthesis was completed 65 years ago, the field remains vibrant with countless players distributed worldwide. Academic syntheses, however, are primarily instructional.
The drug loaded mesoporous hollow monetite exhibited a pH-responsive release behavior. Have great potential in both contaminants adsorbents and drug carriers of orthopedics. ... Synthesis and applications in superior adsorbents for lead ions and pH-responsive release drug carrier Guo, Haifeng; Hu, Siru; Wang, Zongli ...